

Revealing nature's synthetic potential through the study of ribosomal natural product biosynthesis
- PMID: 23286465
- PMCID: PMC3600061
- DOI: 10.1021/cb3005325
Revealing nature's synthetic potential through the study of ribosomal natural product biosynthesis
Erratum in
- ACS Chem Biol. 2013 May 17;8(5):1083
Abstract
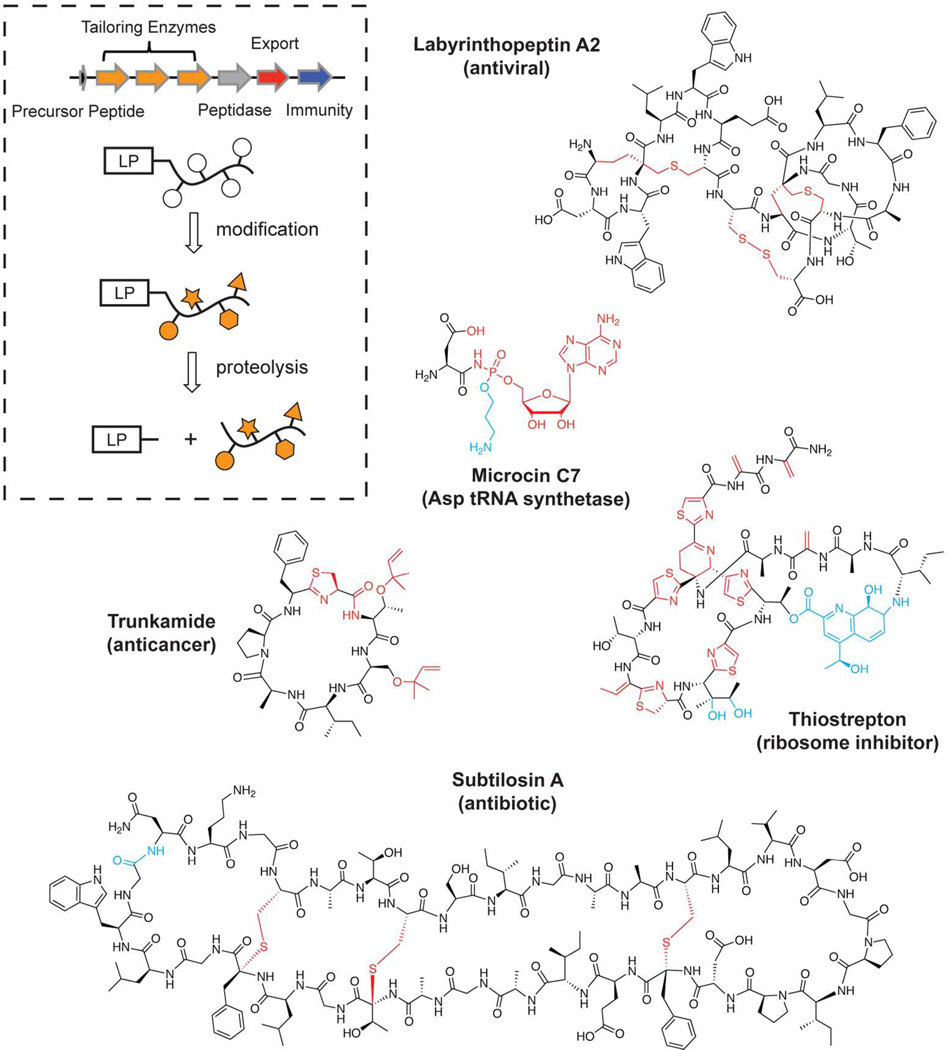
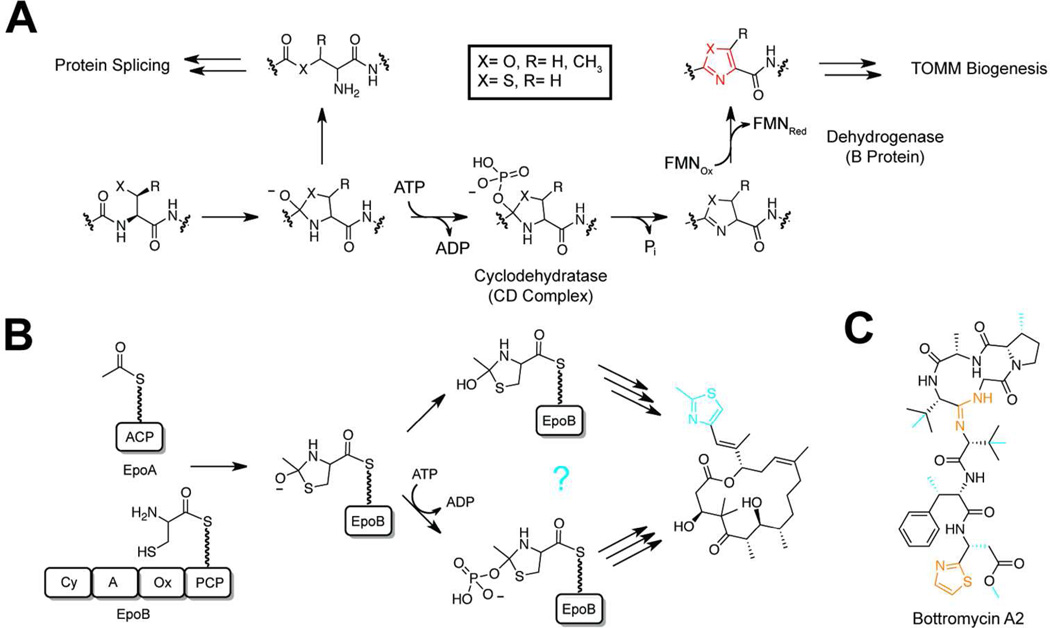
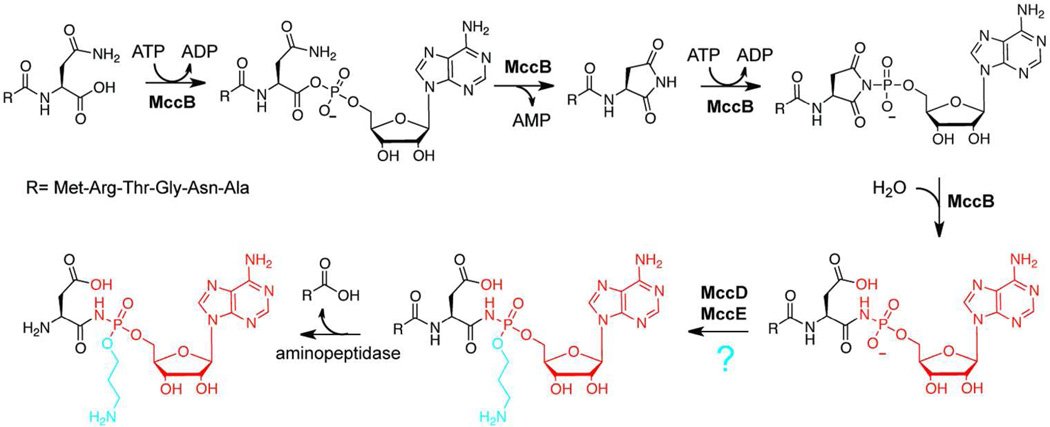
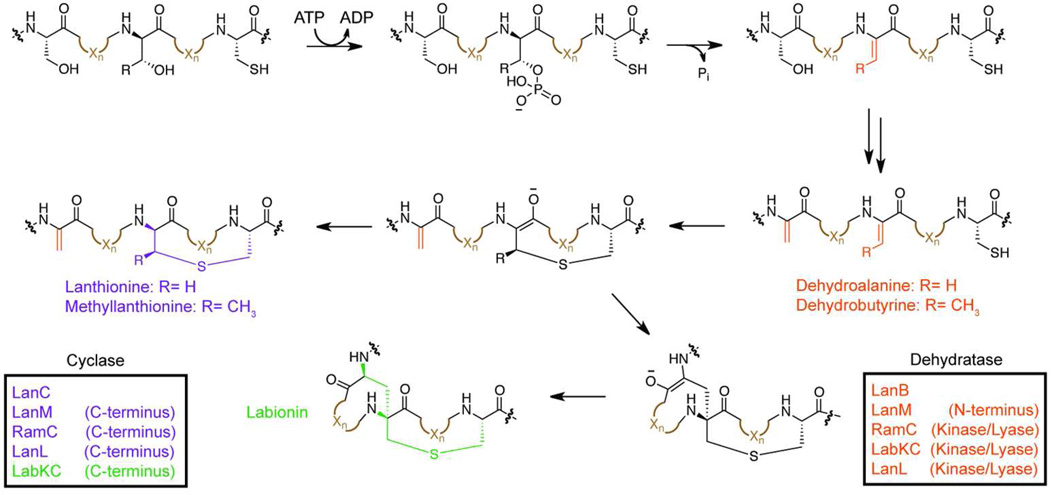
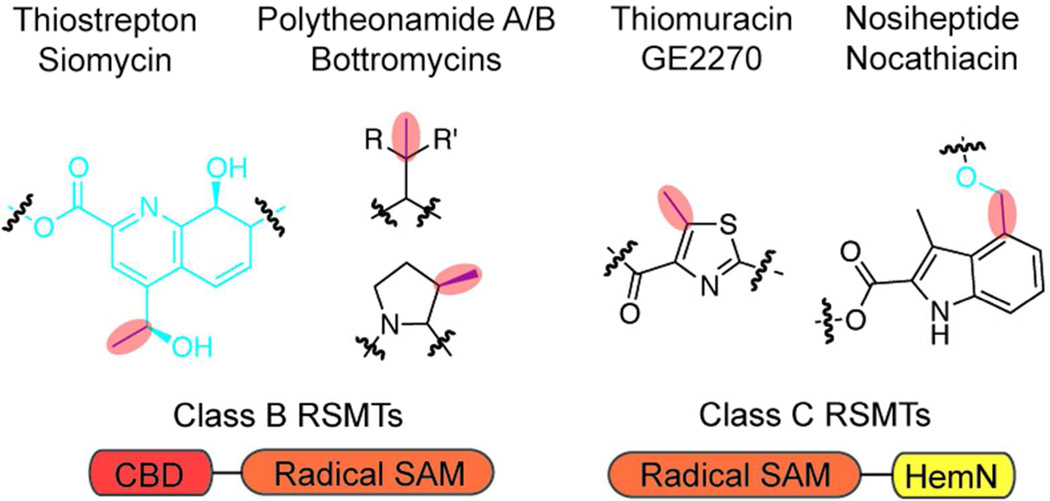
Ribosomally synthesized posttranslationally modified peptides (RiPPs) are a rapidly growing class of natural products with diverse structures and activities. In recent years, a great deal of progress has been made in elucidating the biosynthesis of various RiPP family members. As with the study of nonribosomal peptide and polyketide biosynthetic enzymes, these investigations have led to the discovery of entirely new biological chemistry. With each unique enzyme investigated, a more complex picture of Nature's synthetic potential is revealed. This Review focuses on recent reports (since 2008) that have changed the way that we think about ribosomal natural product biosynthesis and the enzymology of complex bond-forming reactions.
Figures
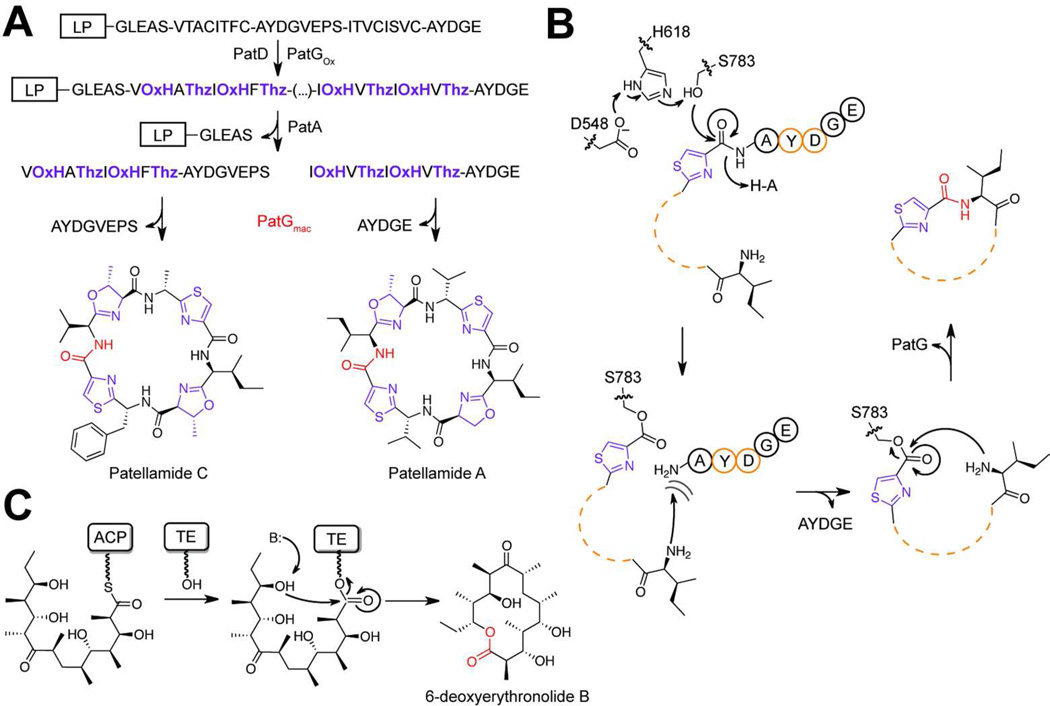
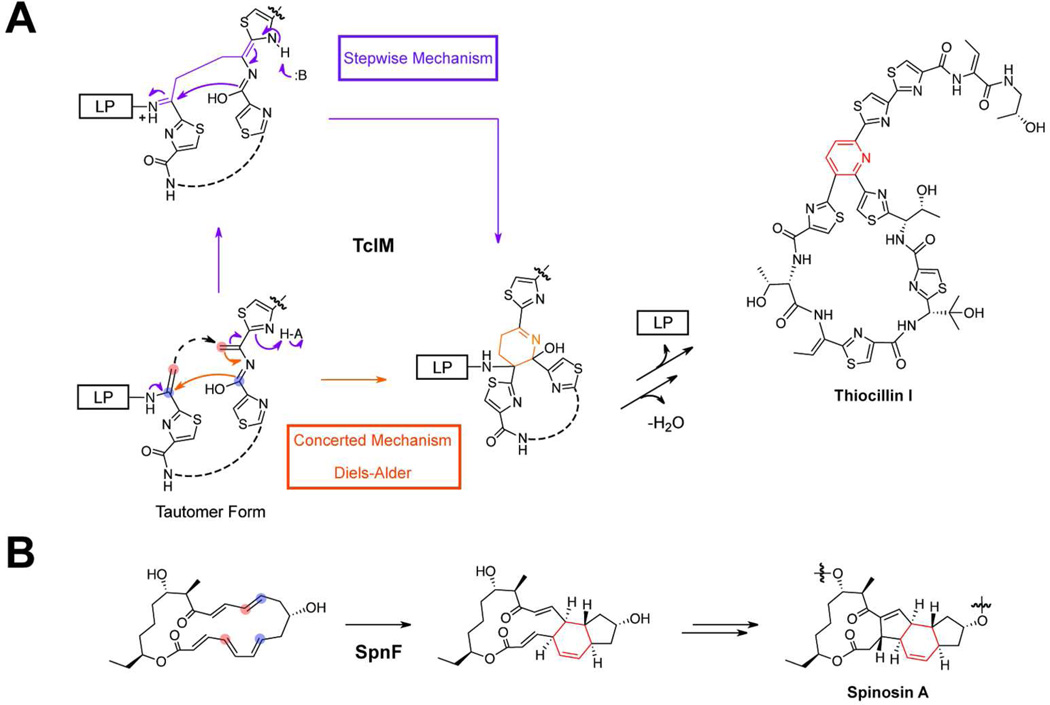
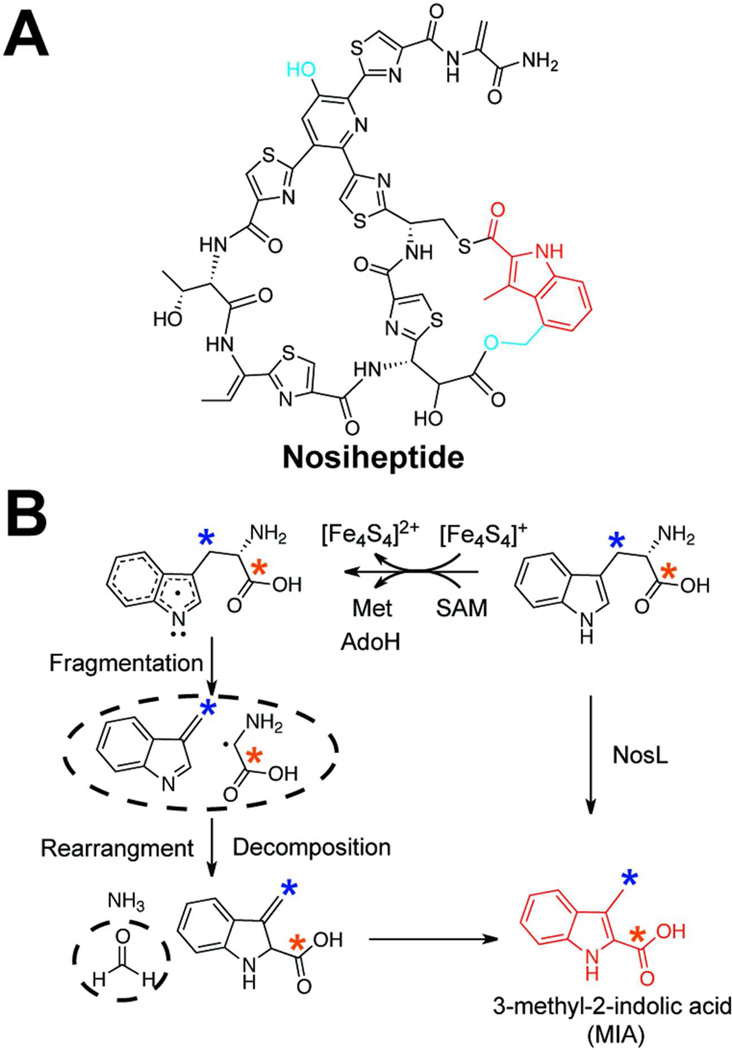
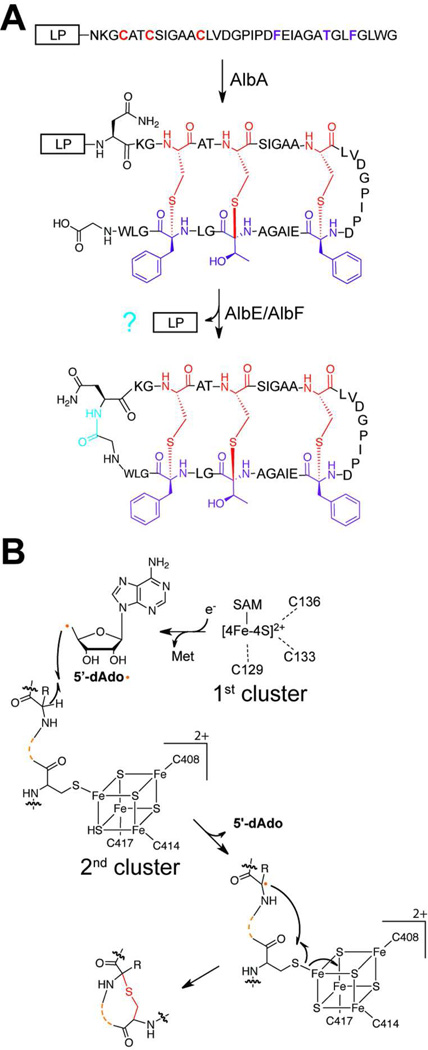



References
-
- Harvey AL. Natural products in drug discovery. Drug Discov. Today. 2008;13:894–901. - PubMed
-
- Wilson RM, Danishefsky SJ. Small molecule natural products in the discovery of therapeutic agents: the synthesis connection. J. Org. Chem. 2006;71:8329–8351. - PubMed
-
- Razzak M, De Brabander JK. Lessons and revelations from biomimetic syntheses. Nat. Chem. Biol. 2011;7:865–875. - PubMed
-
- Falconer SB, Czarny TL, Brown ED. Antibiotics as probes of biological complexity. Nat. Chem. Biol. 2011;7:415–423. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
