

Genomic analysis of smooth tubercle bacilli provides insights into ancestry and pathoadaptation of Mycobacterium tuberculosis
- PMID: 23291586
- PMCID: PMC3856870
- DOI: 10.1038/ng.2517
Genomic analysis of smooth tubercle bacilli provides insights into ancestry and pathoadaptation of Mycobacterium tuberculosis
Abstract
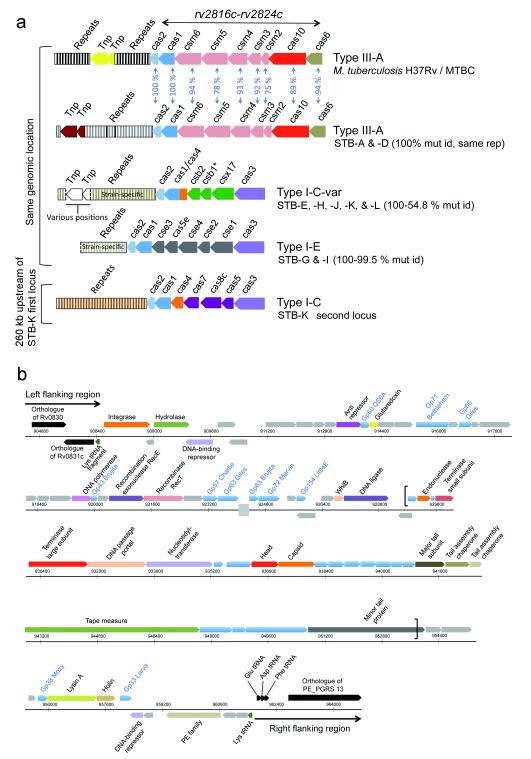
Global spread and limited genetic variation are hallmarks of M. tuberculosis, the agent of human tuberculosis. In contrast, Mycobacterium canettii and related tubercle bacilli that also cause human tuberculosis and exhibit unusual smooth colony morphology are restricted to East Africa. Here, we sequenced and analyzed the whole genomes of five representative strains of smooth tubercle bacilli (STB) using Sanger (4-5× coverage), 454/Roche (13-18× coverage) and/or Illumina DNA sequencing (45-105× coverage). We show that STB isolates are highly recombinogenic and evolutionarily early branching, with larger genome sizes, higher rates of genetic variation, fewer molecular scars and distinct CRISPR-Cas systems relative to M. tuberculosis. Despite the differences, all tuberculosis-causing mycobacteria share a highly conserved core genome. Mouse infection experiments showed that STB strains are less persistent and virulent than M. tuberculosis. We conclude that M. tuberculosis emerged from an ancestral STB-like pool of mycobacteria by gain of persistence and virulence mechanisms, and we provide insights into the molecular events involved.
Figures



References
-
- Dye C, Williams BG. The population dynamics and control of tuberculosis. Science. 2010;328:856–861. - PubMed
-
- Supply P, et al. Linkage disequilibrium between minisatellite loci supports clonal evolution of Mycobacterium tuberculosis in a high tuberculosis incidence area. Mol Microbiol. 2003;47:529–538. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
