

doi: 10.1021/ja3094313.
Epub 2013 Jan 16.
The revolution and evolution of shotgun proteomics for large-scale proteome analysis
Affiliations
- PMID: 23294060
- PMCID: PMC3751590
- DOI: 10.1021/ja3094313
Item in Clipboard
The revolution and evolution of shotgun proteomics for large-scale proteome analysis
J Am Chem Soc.
.
Abstract
Mass spectrometry has evolved at an exponential rate over the last 100 years. Innovations in the development of mass spectrometers have created powerful instruments capable of analyzing a wide range of targets, from rare atoms and molecules to very large molecules, such as a proteins, protein complexes, and DNA. These performance gains have been driven by sustaining innovations, punctuated by the occasional disruptive innovation. The use of mass spectrometry for proteome analysis was driven by disruptive innovations that created a capability for large-scale analysis of proteins and modifications.
Figures
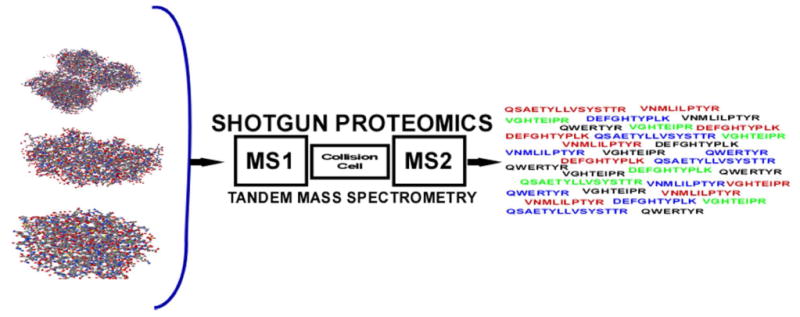
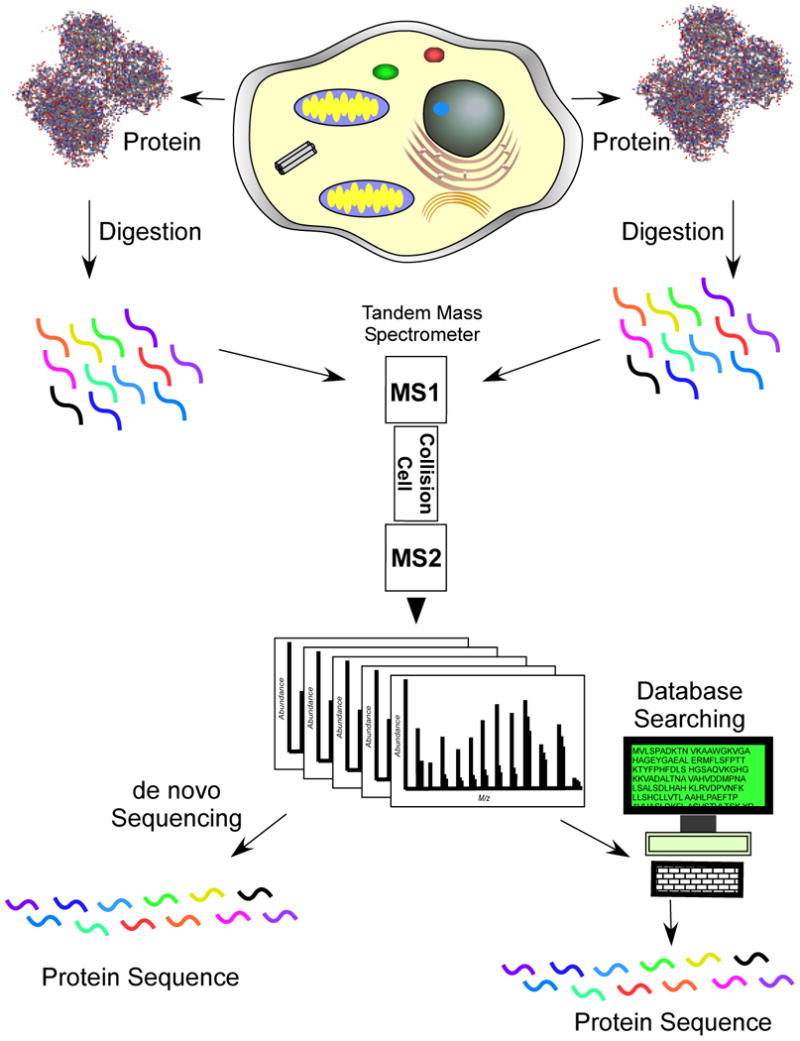
Protein sequencing becomes protein identification. In both strategies the intact protein is digested with a protease, typically trypsin which cleaves after arginine or lysine, to produce a collection of peptides. By using liquid separations coupled to a tandem mass spectrometer, peptide ions are fragmented as they elute into the instrument. In protein sequencing the tandem mass spectrum is interpreted to determine the amino acid sequence de novo. In protein identification, the tandem mass spectrum is searched through a collection of protein sequences to find the best amino acid sequence match to the spectrum.
Shotgun proteomics can be used to directly identify the components of cells, organelles, protein complexes and proteins. Shotgun proteomics also simplifies the analysis of membrane proteins since the proteins can be digested directly in a lipid bilayer rather than trying to enrich the proteins. Membrane proteins are hydrophobic and difficult to manipulate in aqueous buffers.
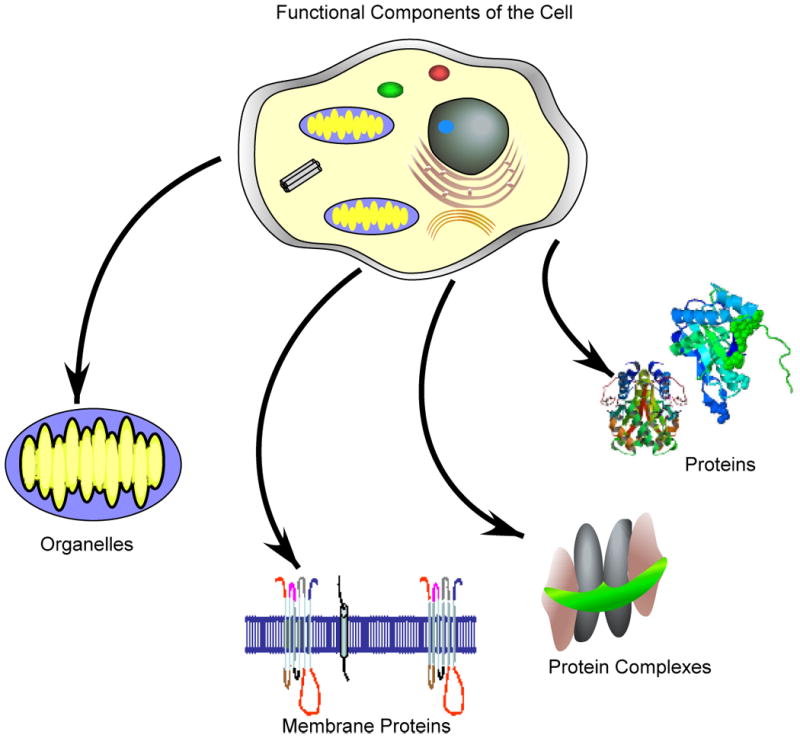
Shotgun proteomics can be used to directly identify the components of cells, organelles, protein complexes and proteins. Shotgun proteomics also simplifies the analysis of membrane proteins since the proteins can be digested directly in a lipid bilayer rather than trying to enrich the proteins. Membrane proteins are hydrophobic and difficult to manipulate in aqueous buffers.
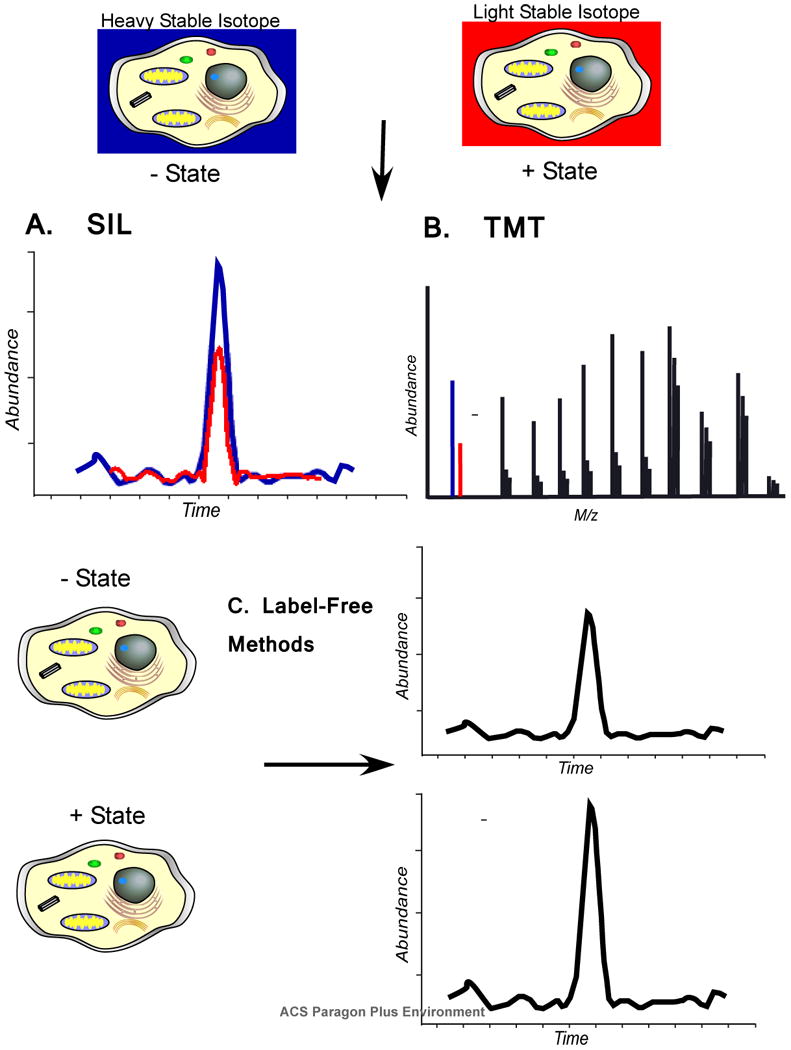
Quantitation of proteins can be performed using stable isotope labels, covalent tags, or label free methods. A) In stable isotope labeling (SIL) methods, a heavy stable isotope label allows peptide masses to be distinguished in the mass spectrometer between different experimental states. Abundance differences can be visualized by selected ion chromatograms. B) Isobaric tags add a mass to the peptides of each state that is isobaric until the peptide ions are fragmented which then reveals a mass difference. The difference in abundance is quantified from the reporter ions in the tandem mass spectrum. C) Two different experimental states can be compared using “label-free” methods. Ion intensity can be measured and compared by selected ion chromatograms as with SIL methods, but these measurements are taken from two different analyses. Another method uses “spectral counting” as a surrogate for abundance based on the observation that proteins which are more abundant have more peptide ions acquired. Label free methods are typically not as accurate as other methods, but can often provide sufficient information to prioritize follow up experiments.
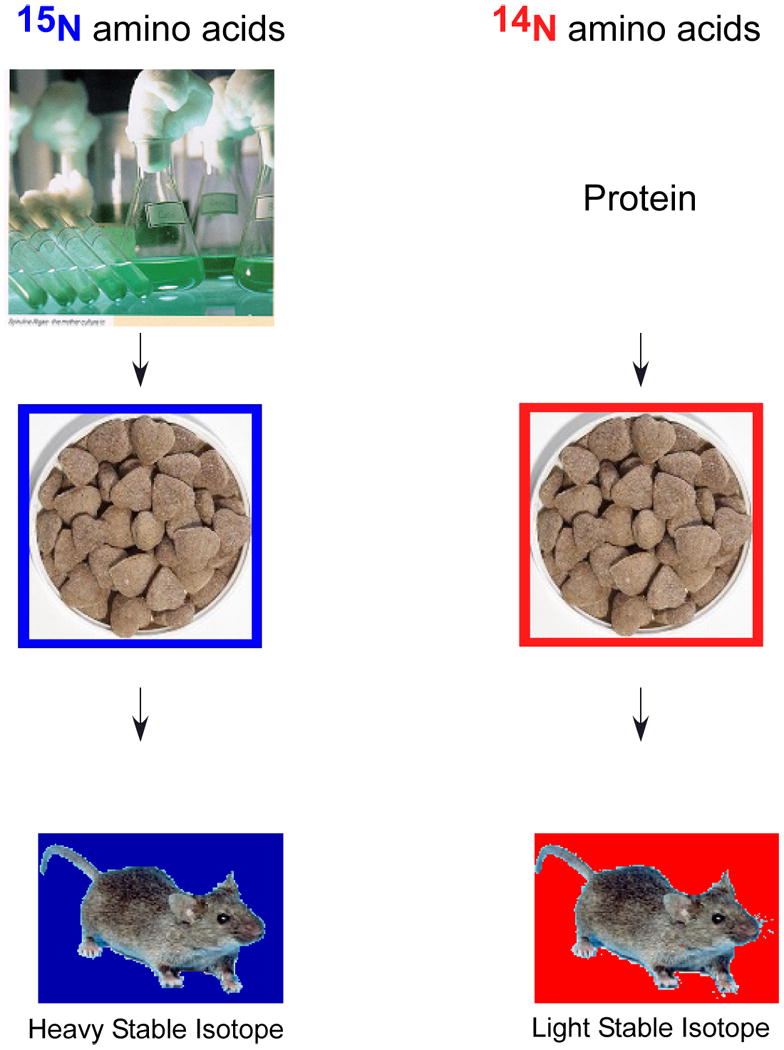
Mice and rats can be labeled with heavy stable isotopes by controlling the protein source in their food. By adding protein labeled with heavy and light stable isotopes to protein free chow, the source of protein in the diet of the animals is restricted to the heavy or light isotopes. These amino acids are incorporated into the new proteins metabolically synthesized by the cells of the animal.
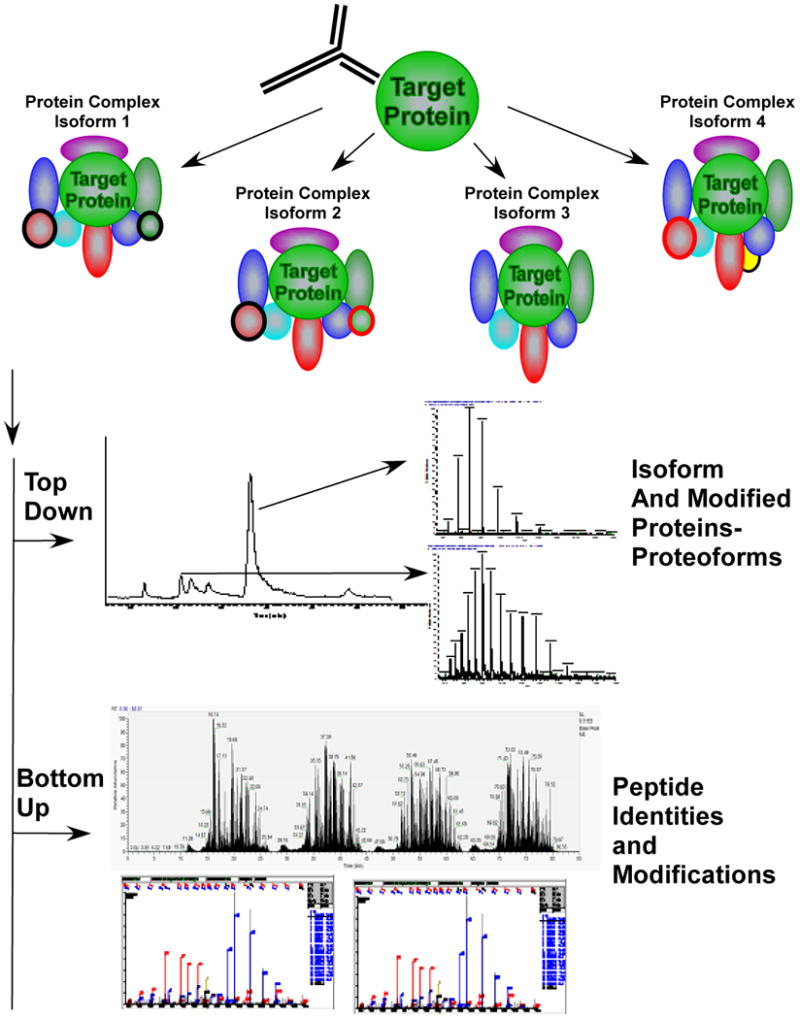
A future challenge to proteomics is deciphering the structures of protein complexes where different proteins, be they isoforms or modified forms may associate at different times or in different locations within a cell with a specific “core” protein. The core protein is depicted as the “target protein” of the enrichment process. To fully characterize the complexes associating with a specific target protein will require methods to separate or enrich the individual complexes, methods such as top down or native mass spectrometry of the complexes or components of the complexes. This data can identify modification status or protein isoforms (proteoforms) present in the complexes. Bottom-up mass spectrometry can accurately identify the proteins present in the complex and modification sites.
References
-
- Malcolm AD. Nature. 1978;275:90. - PubMed
-
- Couzin-Frankel J. Science. 2009;234:1504. - PubMed
-
- Grayson MA. Measuring Mass, From Positive Rays to Proteins. Chemical Heritage Press; Philadelphia: 2002.
-
- Biemann K, Gapp G, Seibl J. J Am Chem Soc. 1959;81:2274.
-
- Biemann K, Lioret C, Asselineau J, Lederer E, Polonsky J. Biochim Biophys Acta. 1960;40:369. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
