

A new locus for X-linked dominant Charcot-Marie-Tooth disease (CMTX6) is caused by mutations in the pyruvate dehydrogenase kinase isoenzyme 3 (PDK3) gene
- PMID: 23297365
- PMCID: PMC3596851
- DOI: 10.1093/hmg/dds557
A new locus for X-linked dominant Charcot-Marie-Tooth disease (CMTX6) is caused by mutations in the pyruvate dehydrogenase kinase isoenzyme 3 (PDK3) gene
Abstract
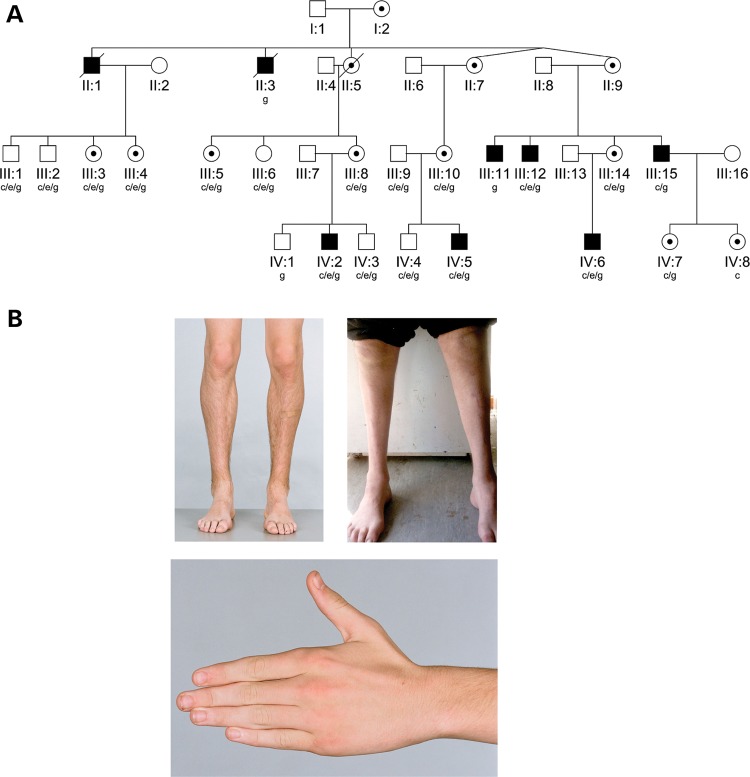
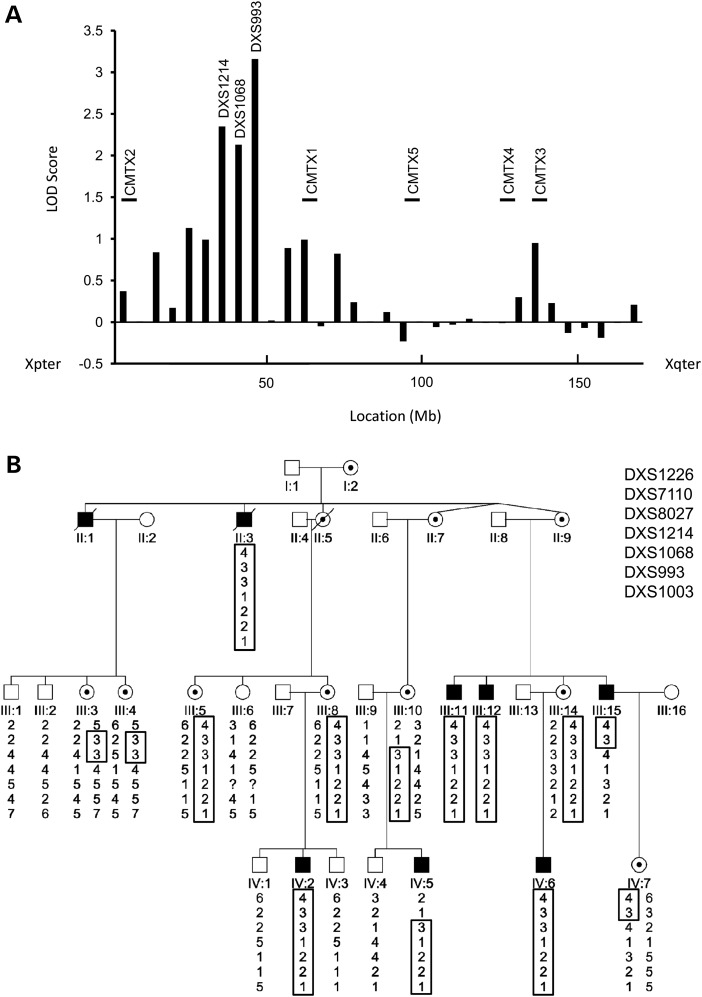
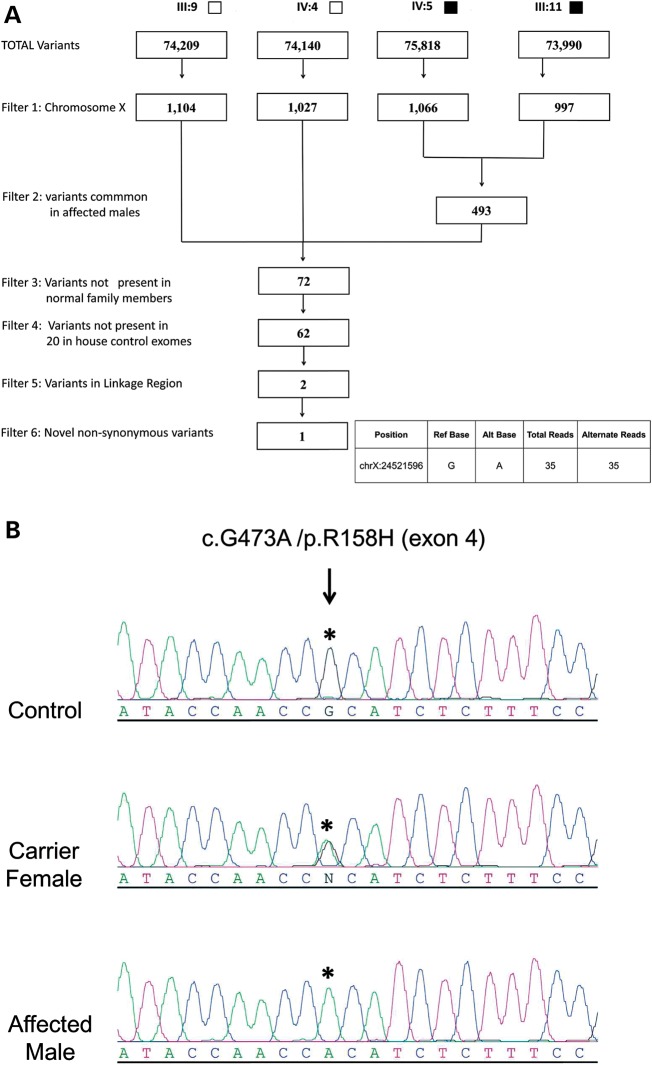
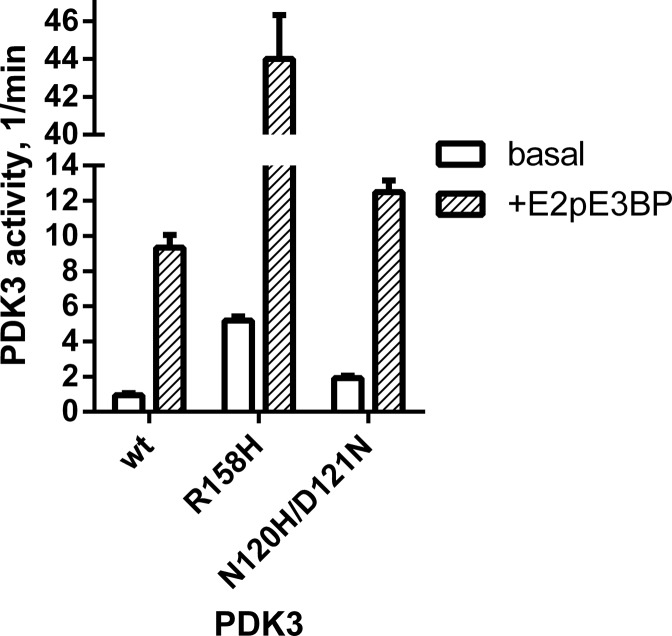
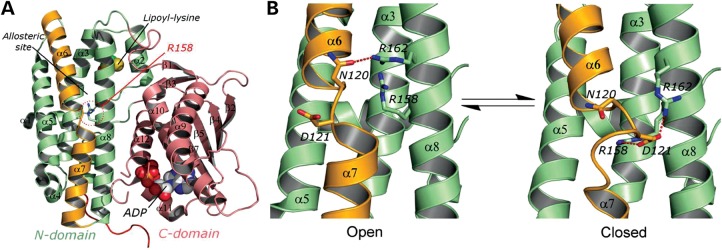
Hereditary motor and sensory disorders of the peripheral nerve form one of the most common groups of human genetic diseases collectively called Charcot-Marie-Tooth (CMT) neuropathy. Using linkage analysis in a three generation kindred, we have mapped a new locus for X-linked dominant CMT to chromosome Xp22.11. A microsatellite scan of the X chromosome established significant linkage to several markers including DXS993 (Zmax = 3.16; θ = 0.05). Extended haplotype analysis refined the linkage region to a 1.43-Mb interval flanked by markers DXS7110 and DXS8027. Whole exome sequencing identified a missense mutation c.G473A (p.R158H) in the pyruvate dehydrogenase kinase isoenzyme 3 (PDK3) gene. The change localized within the 1.43-Mb linkage interval, segregated with the affected phenotype and was excluded in ethnically matched control chromosomes. PDK3 is one of the four isoenzymes regulating the pyruvate dehydrogenase complex (PDC), by reversible phosphorylation, and is a nuclear-coded protein located in the mitochondrial matrix. PDC catalyzes the oxidative decarboxylation of pyruvate to acetyl CoA and is a key enzyme linking glycolysis to the energy-producing Krebs cycle and lipogenic pathways. We found that the R158H mutation confers enzyme hyperactivity and binds with stronger affinity than the wild-type to the inner-lipoyl (L2) domain of the E2p chain of PDC. Our findings suggest a reduced pyruvate flux due to R158H mutant PDK3-mediated hyper-phosphorylation of the PDC as the underlying pathogenic cause of peripheral neuropathy. The results highlight an important causative link between peripheral nerve degeneration and an essential bioenergetic or biosynthetic pathway required for the maintenance of peripheral nerves.
Figures
References
-
- Fowler W.M., Jr., Abresch R.T., Koch T.R., Brewer M.L., Bowden R.K., Wanlass R.L. Employment profiles in neuromuscular diseases. Am. J. Phys. Med. Rehabil. 1997;76:26–37. - PubMed
-
- Bergoffen J., Scherer S.S., Wang S., Scott M.O., Bone L.J., Paul D.L., Chen K., Lensch M.W., Chance P.F., Fischbeck K.H. Connexin mutations in X-linked Charcot-Marie-Tooth disease. Science. 1993;262:2039–2042. - PubMed
-
- Cowchock F.S., Duckett S.W., Streletz L.J., Graziani L.J., Jackson L.G. X-linked motor-sensory neuropathy type-II with deafness and mental retardation: a new disorder. Am. J. Med. Genet. 1985;20:307–315. - PubMed
-
- Priest J.M., Fischbeck K.H., Nouri N., Keats B.J. A locus for axonal motor-sensory neuropathy with deafness and mental retardation maps to Xq24-q26. Genomics. 1995;29:409–412. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
