

Helicobacter pylori-induced disruption of monolayer permeability and proinflammatory cytokine secretion in polarized human gastric epithelial cells
- PMID: 23297384
- PMCID: PMC3584868
- DOI: 10.1128/IAI.01406-12
Helicobacter pylori-induced disruption of monolayer permeability and proinflammatory cytokine secretion in polarized human gastric epithelial cells
Abstract
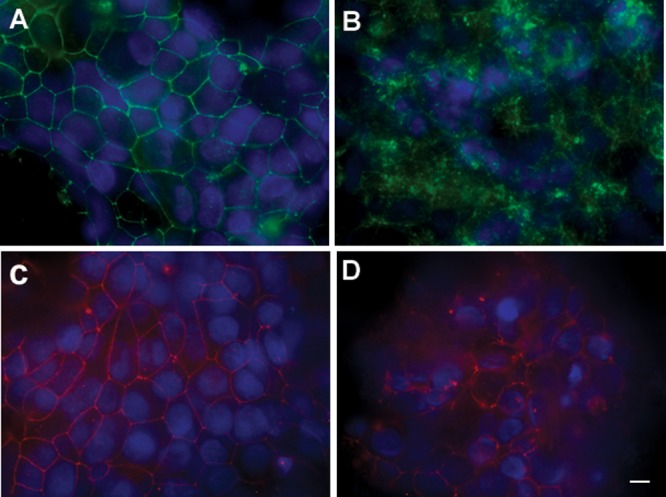
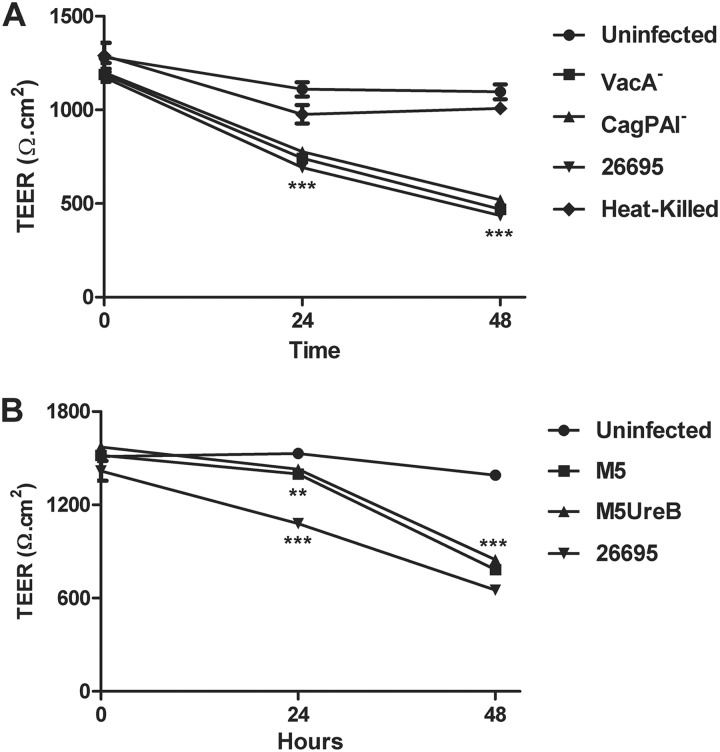
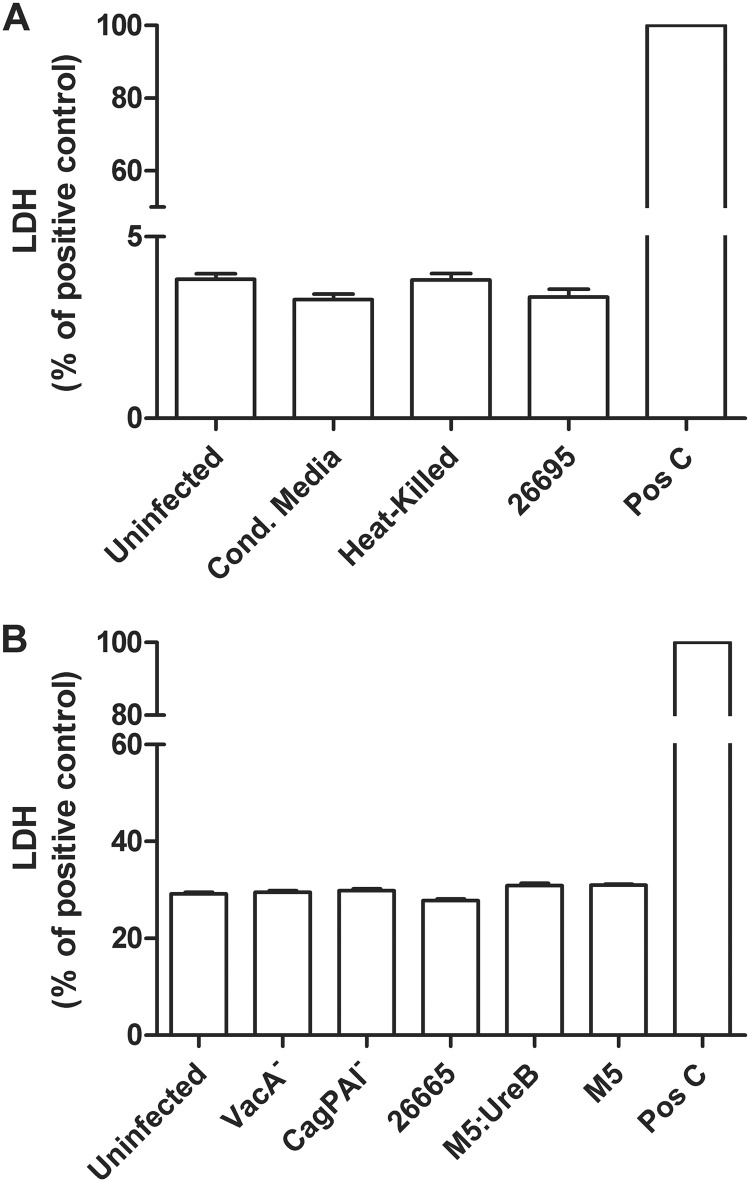
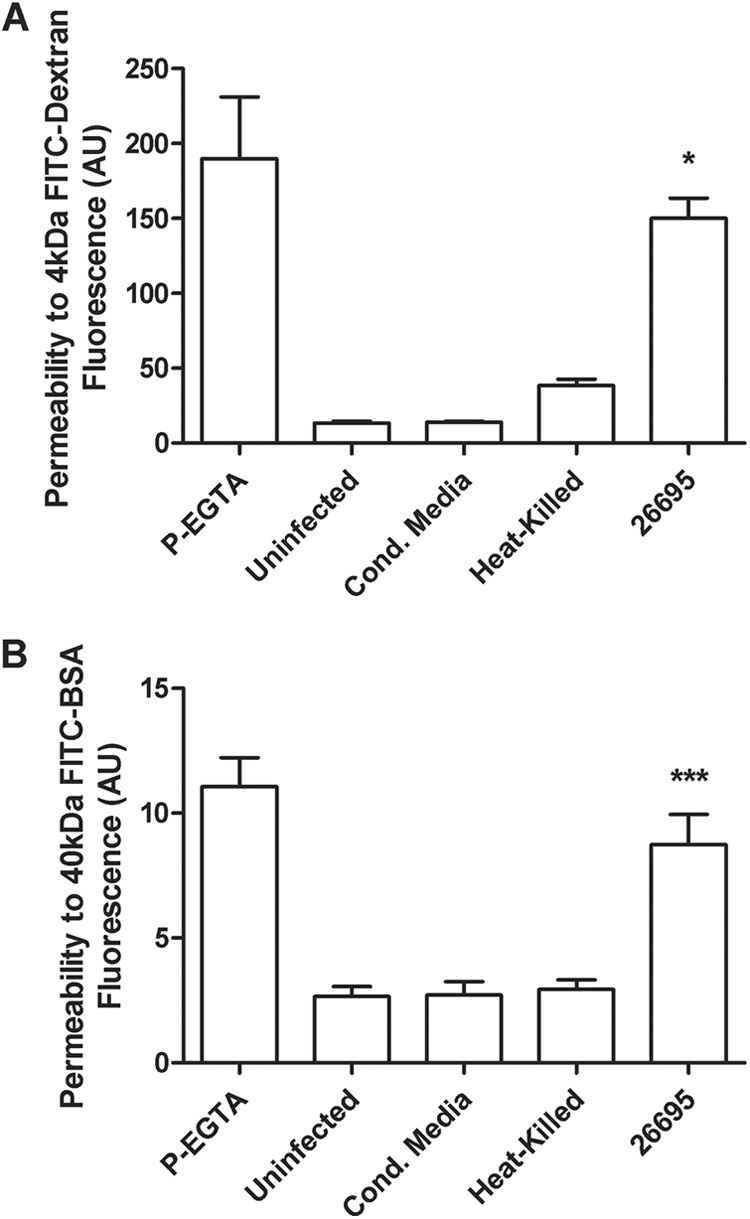
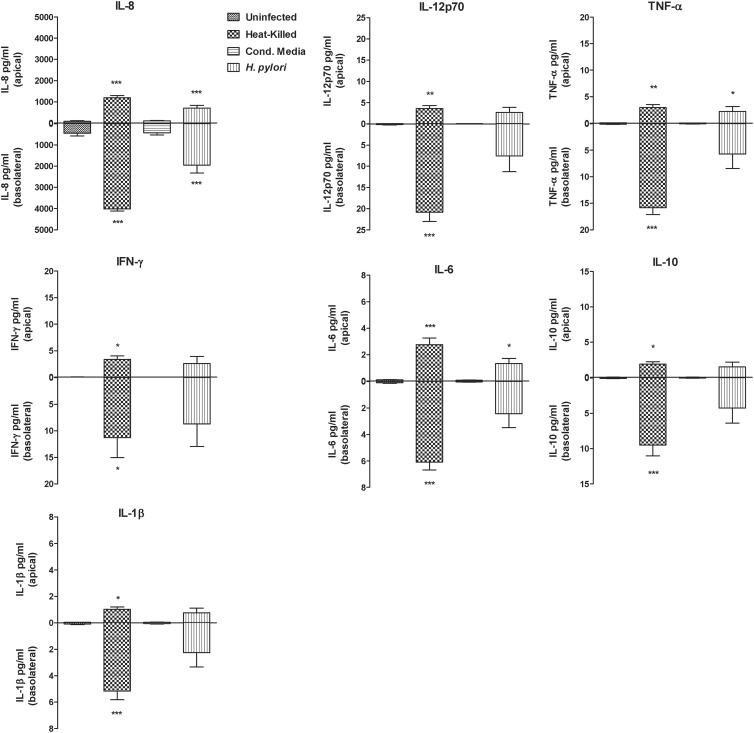
Helicobacter pylori infection of the stomach is related to the development of diverse gastric pathologies. The ability of H. pylori to compromise epithelial junctional complexes and to induce proinflammatory cytokines is believed to contribute to pathogenesis. The purpose of this study was to use an in vitro human gastric epithelial model to investigate the ability of H. pylori to affect permeability and the extent and polarity of the host inflammatory response. NCI-N87 monolayers were cocultured with live or heat-killed H. pylori or culture supernatants. Epithelial barrier function was measured by transepithelial electric resistance (TEER) analysis, diffusion of fluorescein isothiocyanate (FITC)-labeled markers, and immunostaining for tight junction proteins. Supernatants from both apical and basolateral chambers were tested for cytokine production by multiplex analysis. H. pylori caused a significant decrease in TEER, an increased passage of markers through the infected monolayer, and severe disruption and mislocalization of ZO-1 and claudin-1 proteins. Cell viability was not altered by H. pylori, indicating that loss of barrier function could be attributed to a breakdown of tight junction integrity. Significantly high levels of cytokine secretion were induced by either viable or heat-killed H. pylori. H. pylori affects monolayer permeability of polarized human gastric epithelial cells. Proinflammatory cytokines were secreted in a polarized manner, mostly basolaterally. Live bacteria are required for disruption of tight junctions but not for the induction of cytokine secretion. The NCI-N87 cell line provides an excellent model for the in vitro study of H. pylori pathogenesis and the epithelial cell host response to infection.
Figures
References
-
- Marshall BJ, Warren JR. 1984. Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet i: 1311–1315 - PubMed
-
- Everhart JE. 2000. Recent developments in the epidemiology of Helicobacter pylori. Gastroenterol. Clin. North Am. 29: 559–578 - PubMed
-
- NIH Consensus Conference 1994. Helicobacter pylori in peptic ulcer disease. JAMA 272: 65–69 - PubMed
-
- Parsonnet J, Hansen S, Rodriguez L, Gelb AB, Warnke RA, Jellum E, Orentreich N, Vogelman JH, Friedman GD. 1994. Helicobacter pylori infection and gastric lymphoma. N. Engl. J. Med. 330: 1267–1271 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
