

Phosphatidylinositol 4-phosphate 5-kinase α facilitates Toll-like receptor 4-mediated microglial inflammation through regulation of the Toll/interleukin-1 receptor domain-containing adaptor protein (TIRAP) location
- PMID: 23297396
- PMCID: PMC3581370
- DOI: 10.1074/jbc.M112.410126
Phosphatidylinositol 4-phosphate 5-kinase α facilitates Toll-like receptor 4-mediated microglial inflammation through regulation of the Toll/interleukin-1 receptor domain-containing adaptor protein (TIRAP) location
Abstract
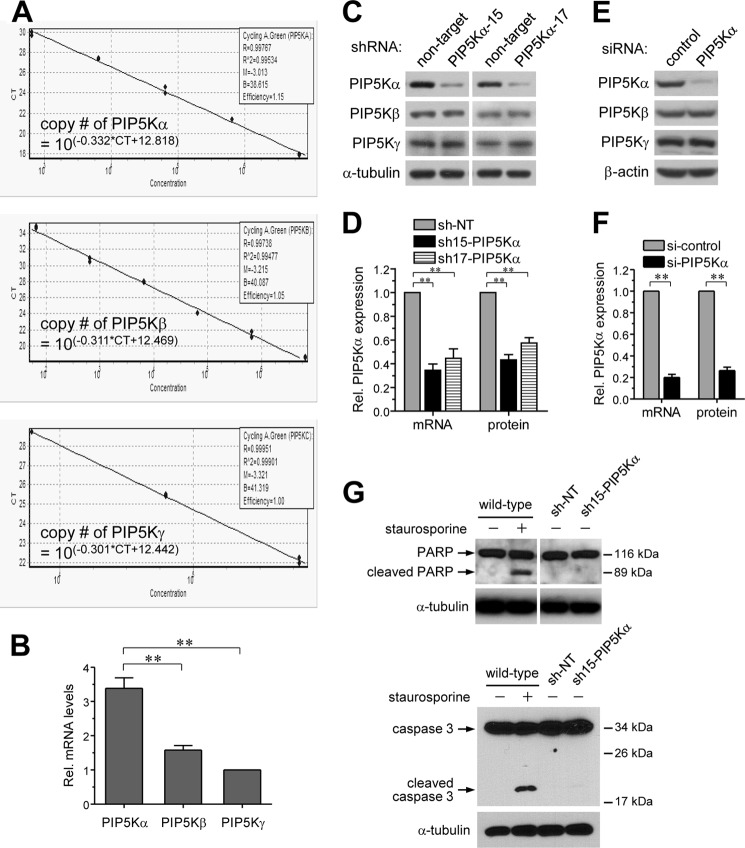
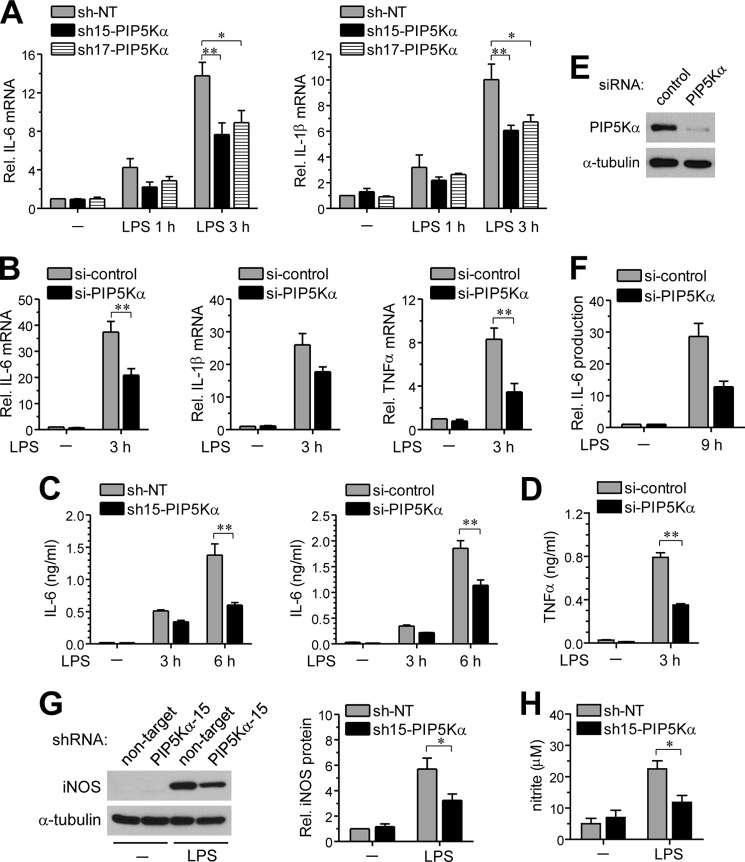
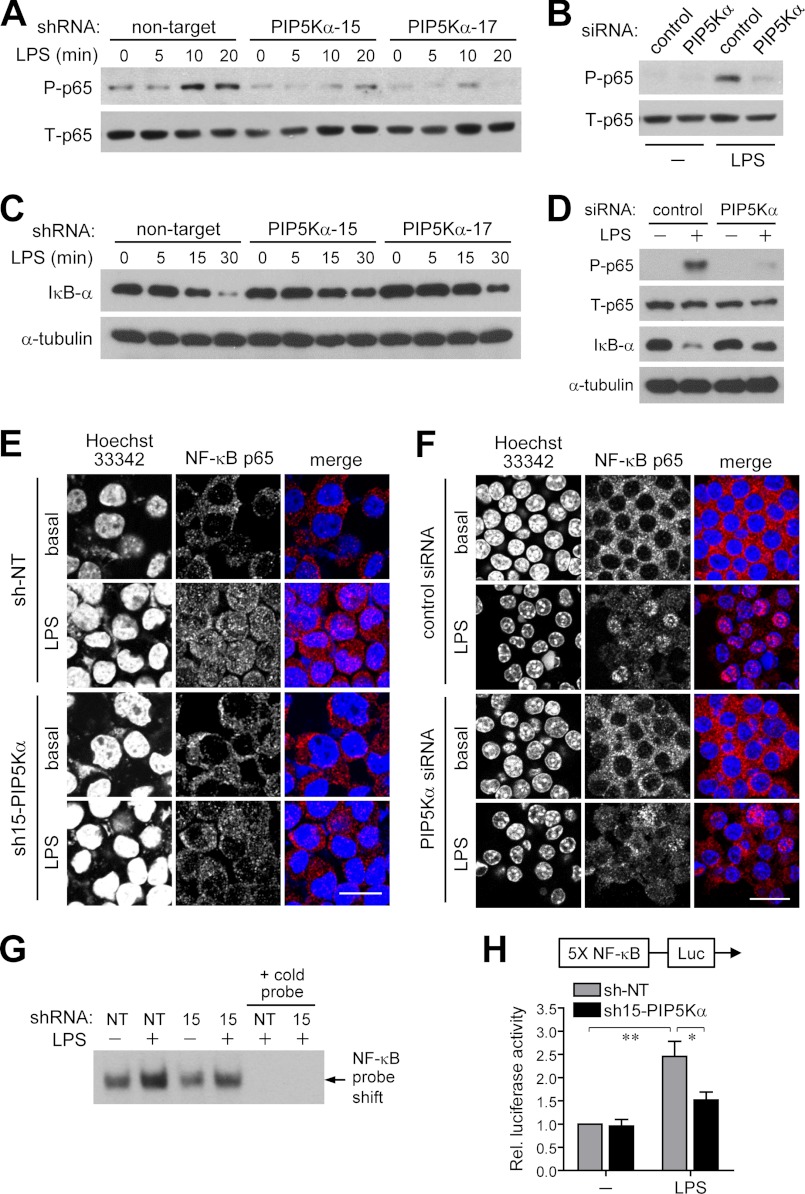
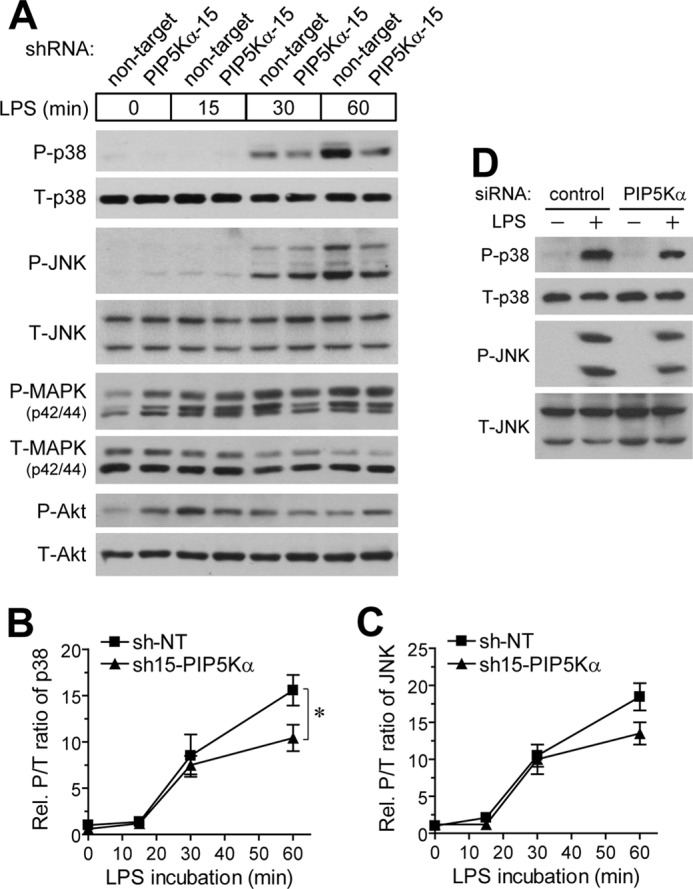
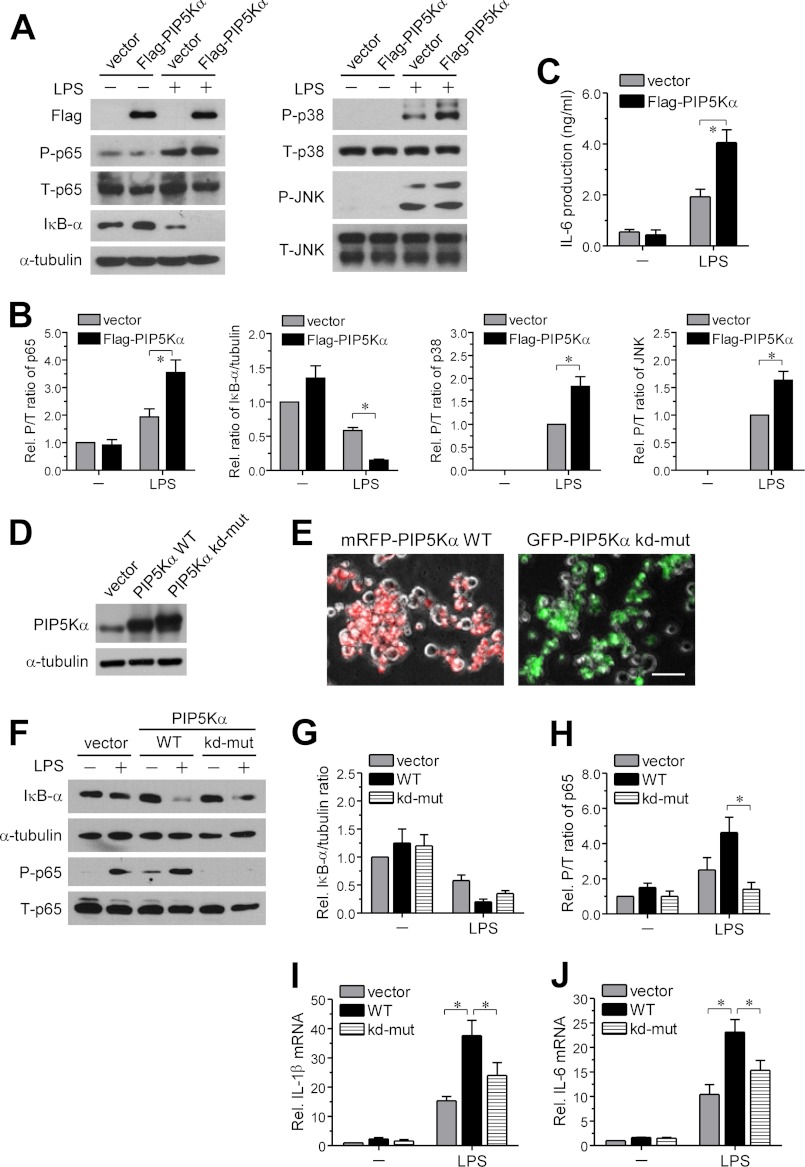
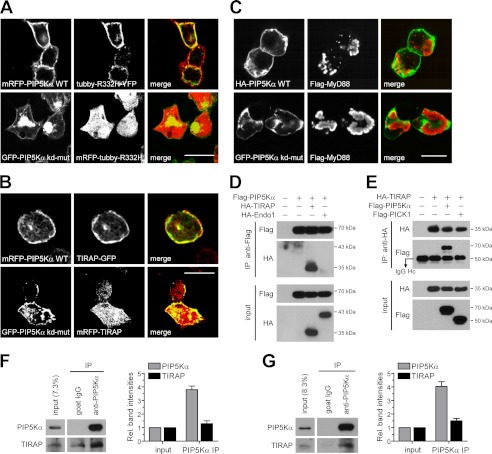
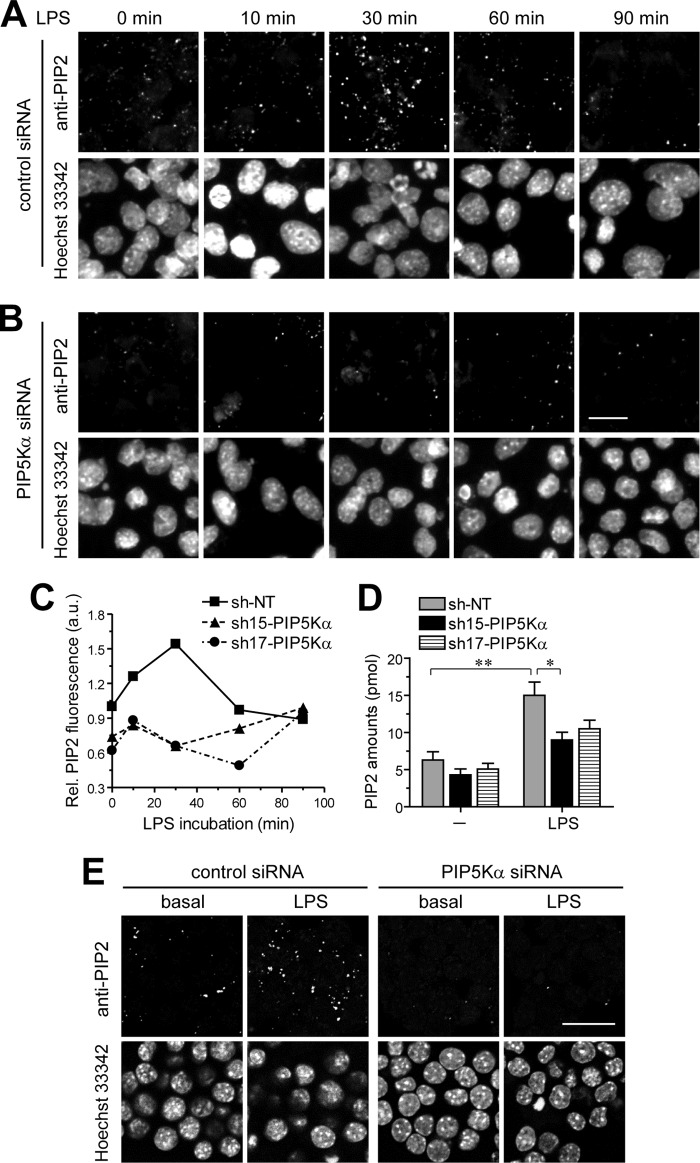
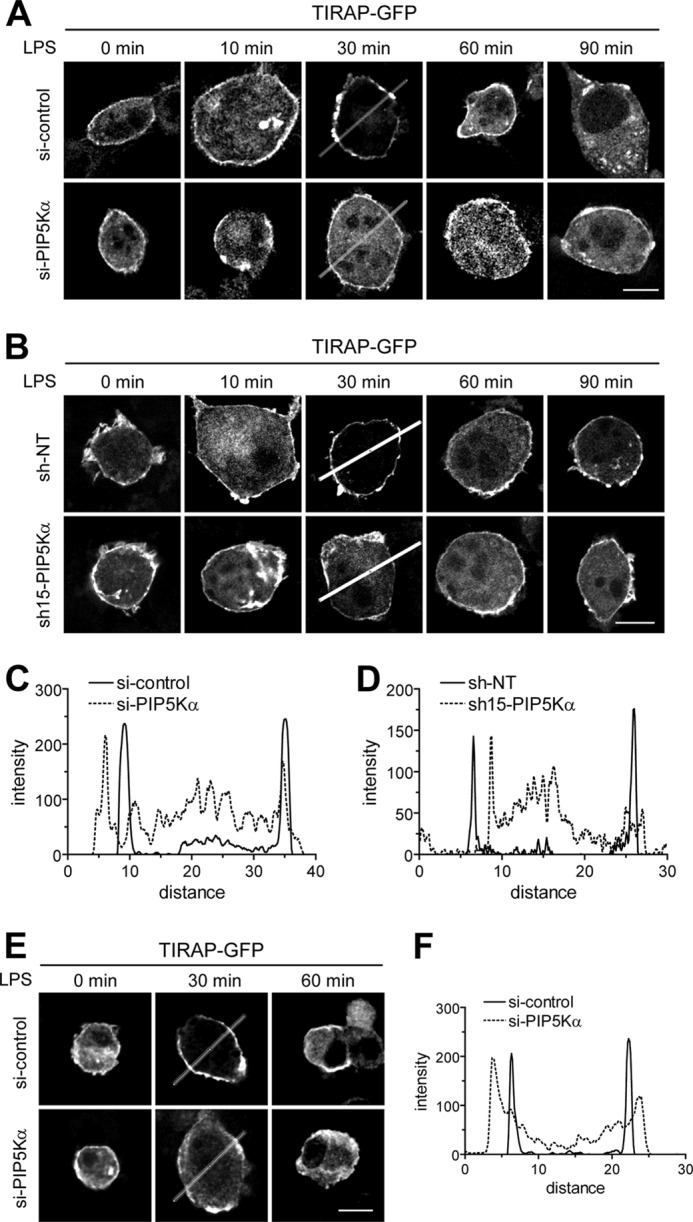
Phosphatidylinositol (PI) 4,5-bisphosphate (PIP(2)), generated by PI 4-phosphate 5-kinase (PIP5K), regulates many critical cellular events. PIP(2) is also known to mediate plasma membrane localization of the Toll/IL-1 receptor domain-containing adaptor protein (TIRAP), required for the MyD88-dependent Toll-like receptor (TLR) 4 signaling pathway. Microglia are the primary immune competent cells in brain tissue, and TLR4 is important for microglial activation. However, a functional role for PIP5K and PIP(2) in TLR4-dependent microglial activation remains unclear. Here, we knocked down PIP5Kα, a PIP5K isoform, in a BV2 microglial cell line using stable expression of lentiviral shRNA constructs or siRNA transfection. PIP5Kα knockdown significantly suppressed induction of inflammatory mediators, including IL-6, IL-1β, and nitric oxide, by lipopolysaccharide. PIP5Kα knockdown also attenuated signaling events downstream of TLR4 activation, including p38 MAPK and JNK phosphorylation, NF-κB p65 nuclear translocation, and IκB-α degradation. Complementation of the PIP5Kα knockdown cells with wild type but not kinase-dead PIP5Kα effectively restored the LPS-mediated inflammatory response. We found that PIP5Kα and TIRAP colocalized at the cell surface and interacted with each other, whereas kinase-dead PIP5Kα rendered TIRAP soluble. Furthermore, in LPS-stimulated control cells, plasma membrane PIP(2) increased and subsequently declined, and TIRAP underwent bi-directional translocation between the membrane and cytosol, which temporally correlated with the changes in PIP(2). In contrast, PIP5Kα knockdown that reduced PIP(2) levels disrupted TIRAP membrane targeting by LPS. Together, our results suggest that PIP5Kα promotes TLR4-associated microglial inflammation by mediating PIP(2)-dependent recruitment of TIRAP to the plasma membrane.
Figures
References
-
- Aloisi F. (2001) Immune function of microglia. Glia 36, 165–179 - PubMed
-
- Hanisch U. K., Kettenmann H. (2007) Microglia. Active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci. 10, 1387–1394 - PubMed
-
- Kim S. U., de Vellis J. (2005) Microglia in health and disease. J. Neurosci. Res. 81, 302–313 - PubMed
-
- Block M. L., Zecca L., Hong J. S. (2007) Microglia-mediated neurotoxicity. Uncovering the molecular mechanisms. Nat. Rev. Neurosci. 8, 57–69 - PubMed
-
- Yang M. S., Min K. J., Joe E. (2007) Multiple mechanisms that prevent excessive brain inflammation. J. Neurosci. Res. 85, 2298–2305 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
