

Upregulated autophagy protects cardiomyocytes from oxidative stress-induced toxicity
- PMID: 23298947
- PMCID: PMC3590254
- DOI: 10.4161/auto.22971
Upregulated autophagy protects cardiomyocytes from oxidative stress-induced toxicity
Abstract
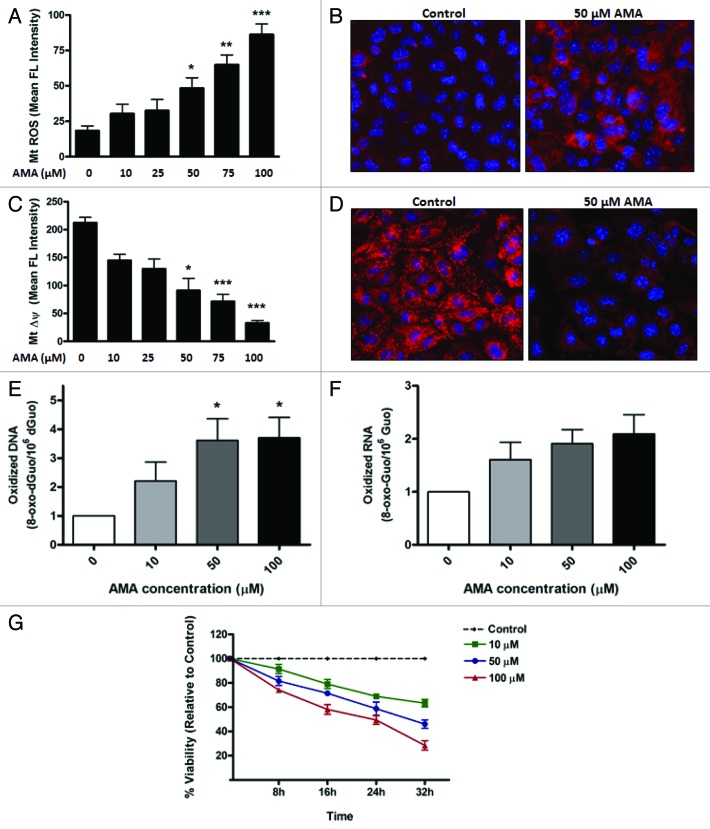
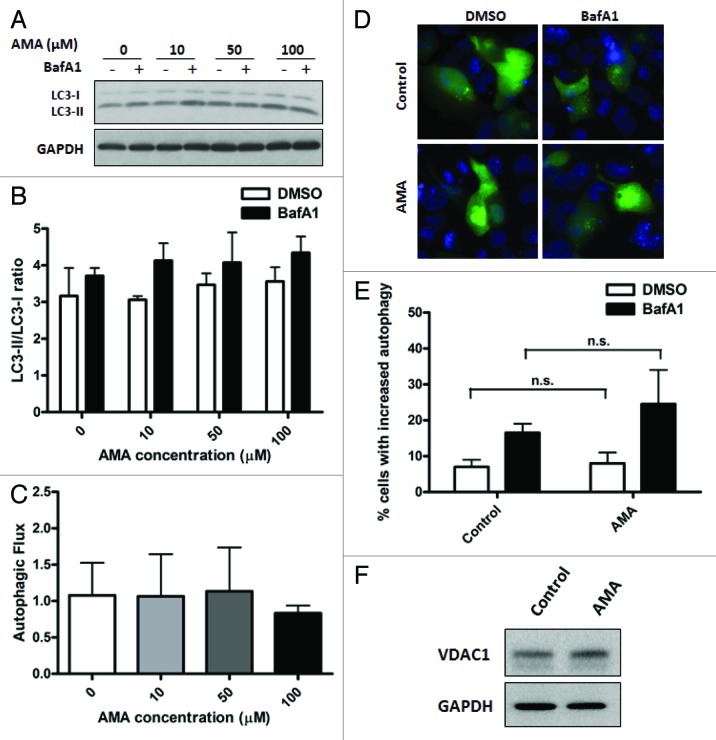
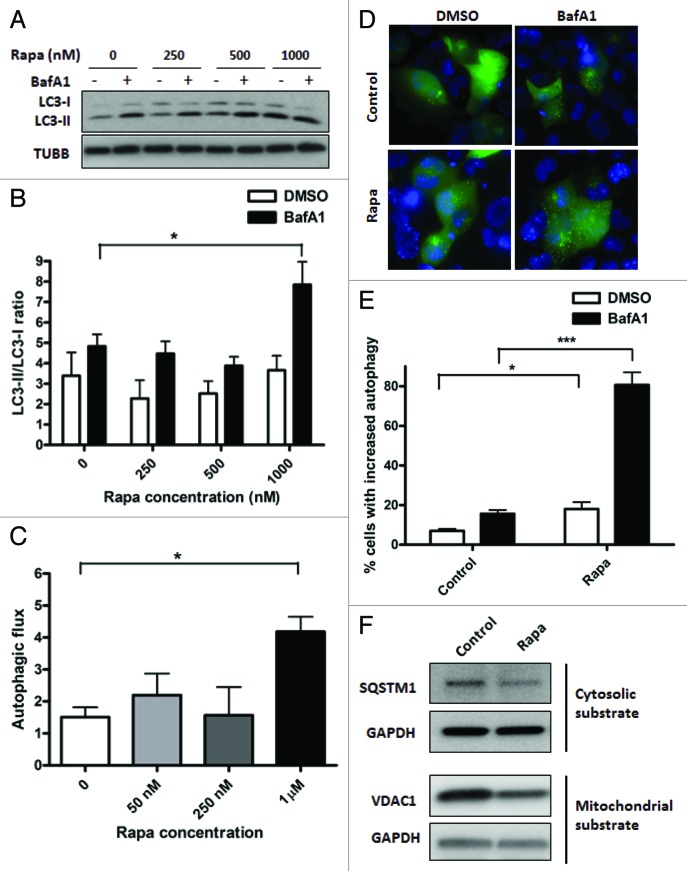
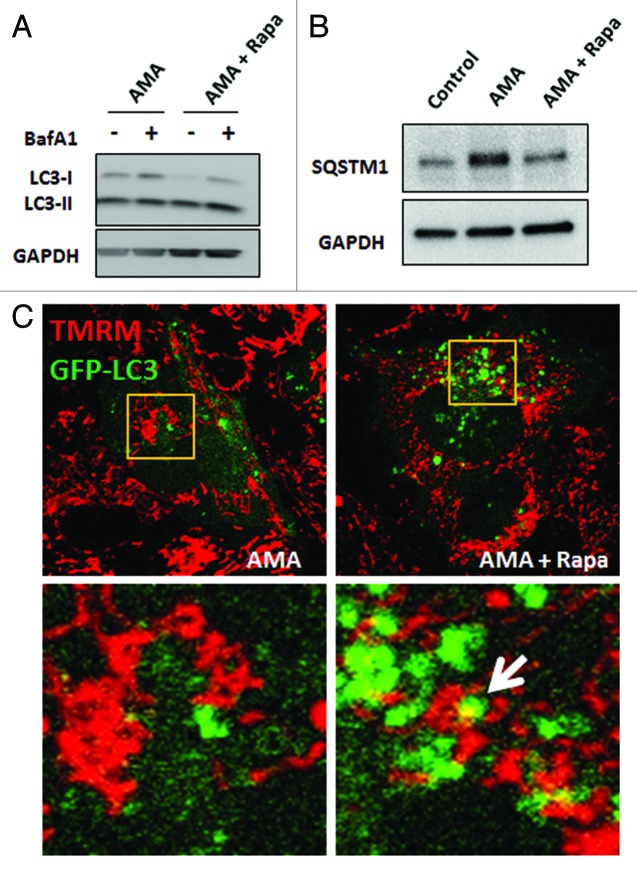
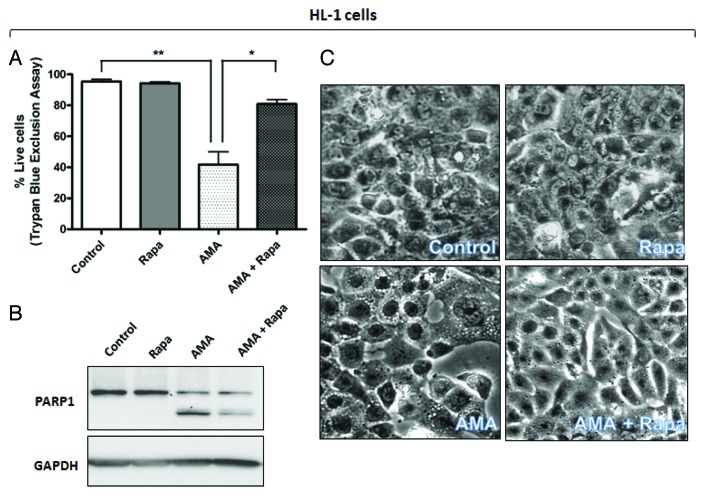
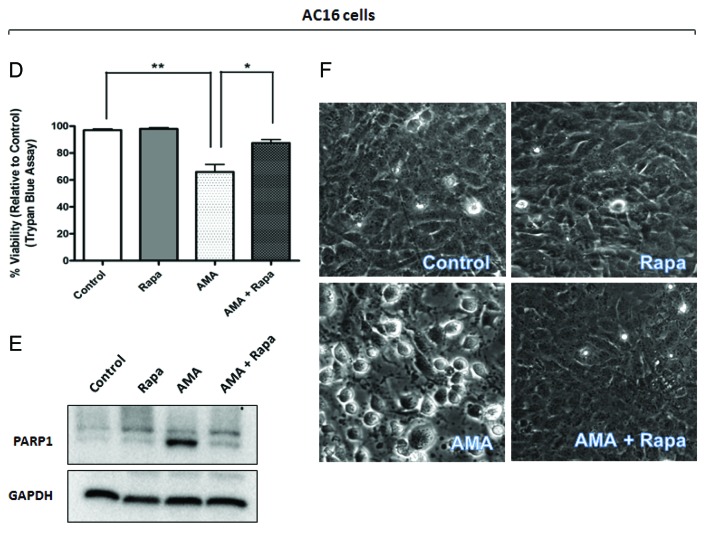
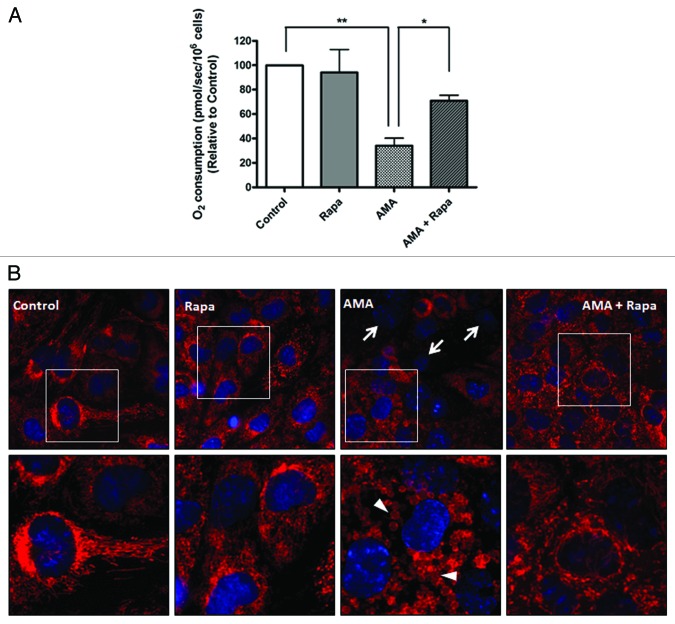
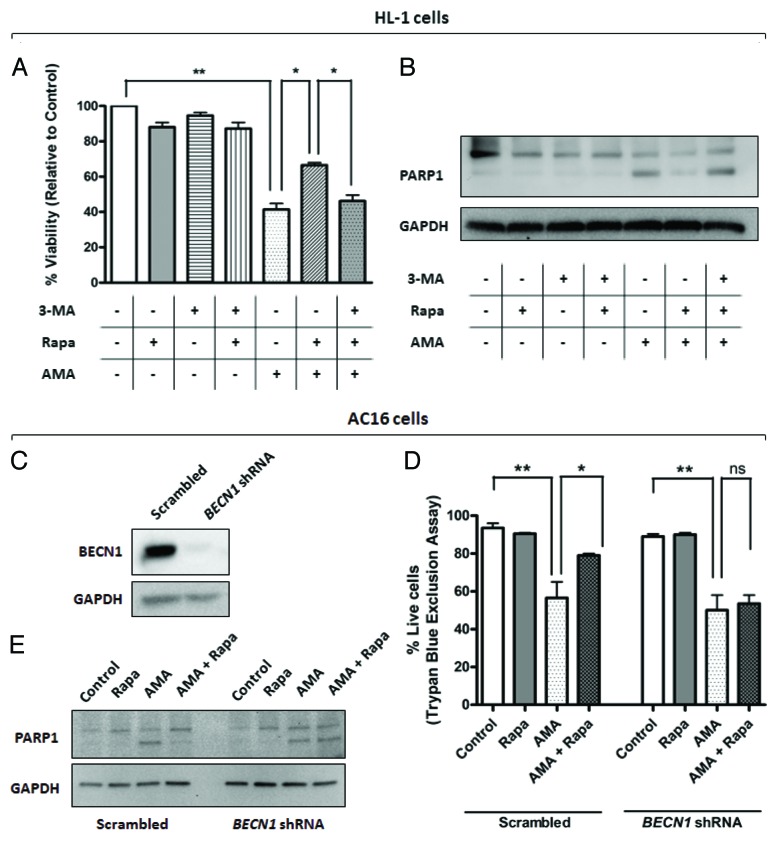
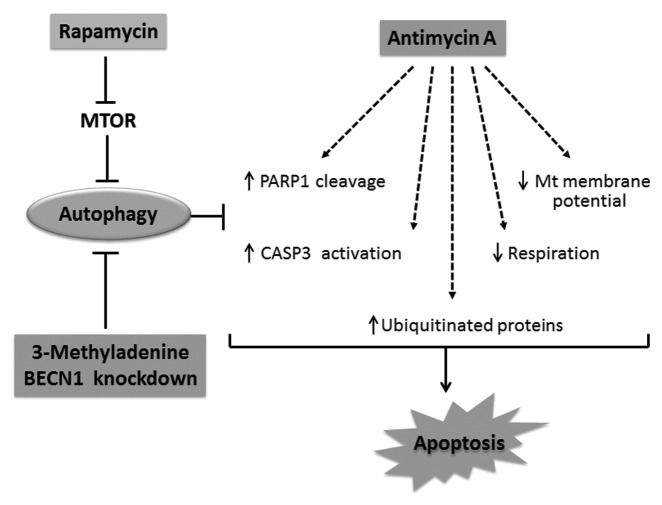
Autophagy is a cellular self-digestion process that mediates protein quality control and serves to protect against neurodegenerative disorders, infections, inflammatory diseases and cancer. Current evidence suggests that autophagy can selectively remove damaged organelles such as the mitochondria. Mitochondria-induced oxidative stress has been shown to play a major role in a wide range of pathologies in several organs, including the heart. Few studies have investigated whether enhanced autophagy can offer protection against mitochondrially-generated oxidative stress. We induced mitochondrial stress in cardiomyocytes using antimycin A (AMA), which increased mitochondrial superoxide generation, decreased mitochondrial membrane potential and depressed cellular respiration. In addition, AMA augmented nuclear DNA oxidation and cell death in cardiomyocytes. Interestingly, although oxidative stress has been proposed to induce autophagy, treatment with AMA did not result in stimulation of autophagy or mitophagy in cardiomyocytes. Our results showed that the MTOR inhibitor rapamycin induced autophagy, promoted mitochondrial clearance and protected cardiomyocytes from the cytotoxic effects of AMA, as assessed by apoptotic marker activation and viability assays in both mouse atrial HL-1 cardiomyocytes and human ventricular AC16 cells. Importantly, rapamycin improved mitochondrial function, as determined by cellular respiration, mitochondrial membrane potential and morphology analysis. Furthermore, autophagy induction by rapamycin suppressed the accumulation of ubiquitinylated proteins induced by AMA. Inhibition of rapamycin-induced autophagy by pharmacological or genetic interventions attenuated the cytoprotective effects of rapamycin against AMA. We propose that rapamycin offers cytoprotection against oxidative stress by a combined approach of removing dysfunctional mitochondria as well as by degrading damaged, ubiquitinated proteins. We conclude that autophagy induction by rapamycin could be utilized as a potential therapeutic strategy against oxidative stress-mediated damage in cardiomyocytes.
Keywords: MTOR; autophagy; cardiomyocytes; mitochondrial dysfunction; oxidative stress; rapamycin.
Figures
References
-
- Judge S, Jang YM, Smith A, Hagen T, Leeuwenburgh C. Age-associated increases in oxidative stress and antioxidant enzyme activities in cardiac interfibrillar mitochondria: implications for the mitochondrial theory of aging. FASEB J. 2005;19:419–21. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous