

A genomic portrait of the emergence, evolution, and global spread of a methicillin-resistant Staphylococcus aureus pandemic
- PMID: 23299977
- PMCID: PMC3613582
- DOI: 10.1101/gr.147710.112
A genomic portrait of the emergence, evolution, and global spread of a methicillin-resistant Staphylococcus aureus pandemic
Abstract
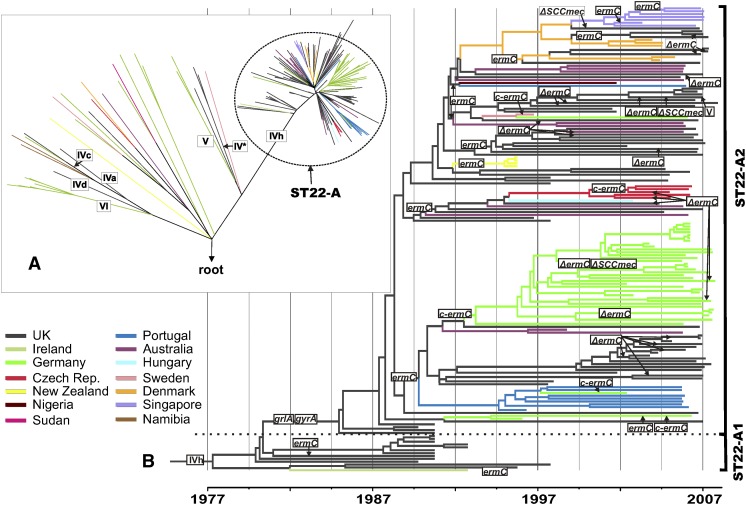
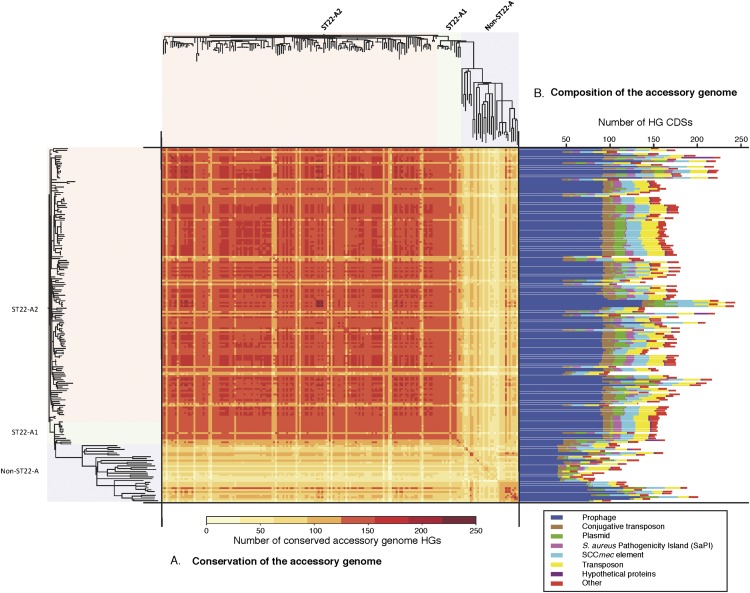
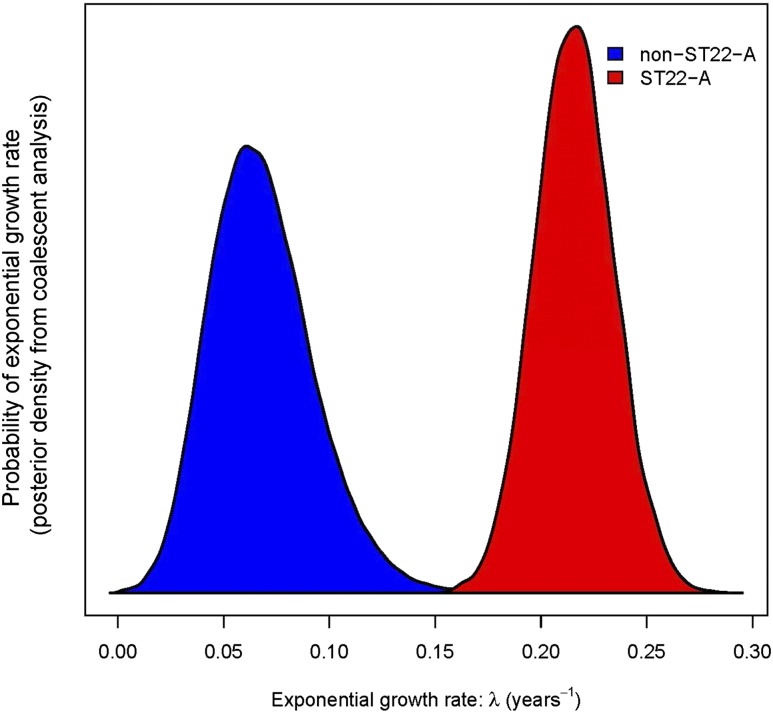
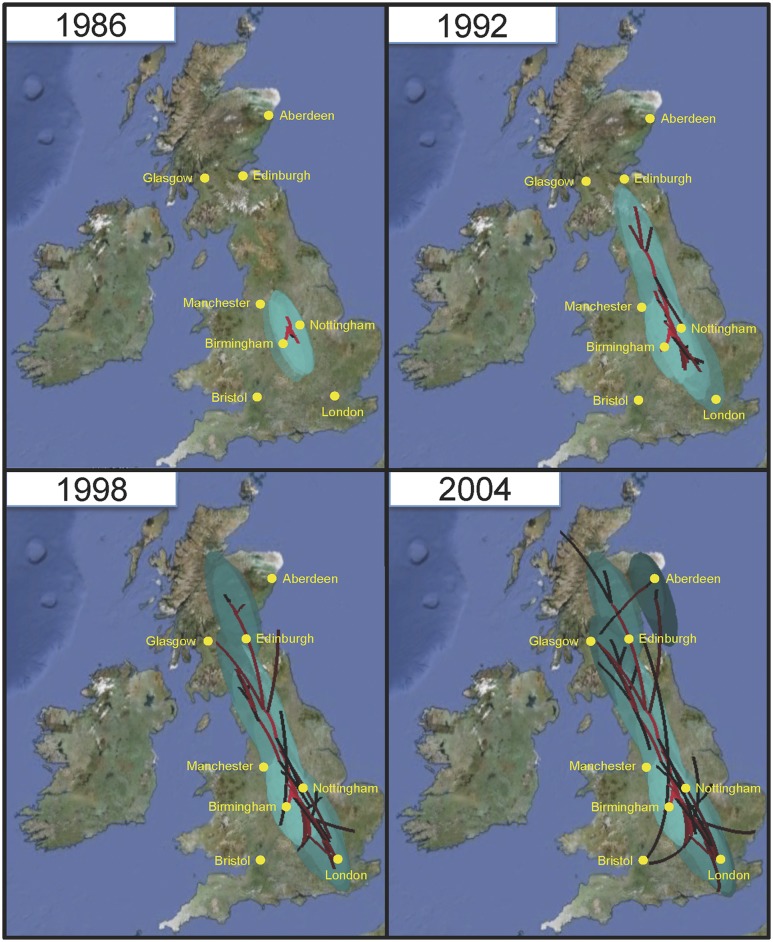
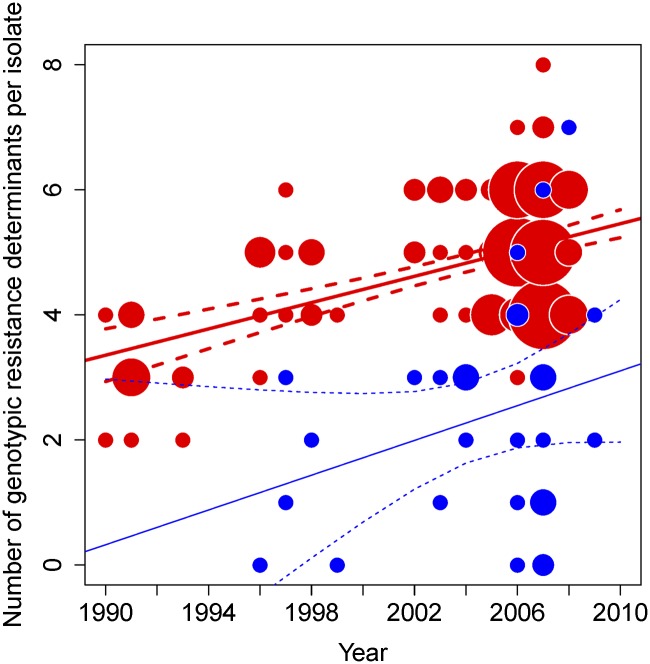
The widespread use of antibiotics in association with high-density clinical care has driven the emergence of drug-resistant bacteria that are adapted to thrive in hospitalized patients. Of particular concern are globally disseminated methicillin-resistant Staphylococcus aureus (MRSA) clones that cause outbreaks and epidemics associated with health care. The most rapidly spreading and tenacious health-care-associated clone in Europe currently is EMRSA-15, which was first detected in the UK in the early 1990s and subsequently spread throughout Europe and beyond. Using phylogenomic methods to analyze the genome sequences for 193 S. aureus isolates, we were able to show that the current pandemic population of EMRSA-15 descends from a health-care-associated MRSA epidemic that spread throughout England in the 1980s, which had itself previously emerged from a primarily community-associated methicillin-sensitive population. The emergence of fluoroquinolone resistance in this EMRSA-15 subclone in the English Midlands during the mid-1980s appears to have played a key role in triggering pandemic spread, and occurred shortly after the first clinical trials of this drug. Genome-based coalescence analysis estimated that the population of this subclone over the last 20 yr has grown four times faster than its progenitor. Using comparative genomic analysis we identified the molecular genetic basis of 99.8% of the antimicrobial resistance phenotypes of the isolates, highlighting the potential of pathogen genome sequencing as a diagnostic tool. We document the genetic changes associated with adaptation to the hospital environment and with increasing drug resistance over time, and how MRSA evolution likely has been influenced by country-specific drug use regimens.
Figures
References
-
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ 1990. Basic local alignment search tool. J Mol Biol 215: 403–410 - PubMed
-
- Amorim ML, Faria NA, Oliveira DC, Vasconcelos C, Cabeda JC, Mendes AC, Calado E, Castro AP, Ramos MH, Amorim JM, et al. 2007. Changes in the clonal nature and antibiotic resistance profiles of methicillin-resistant Staphylococcus aureus isolates associated with spread of the EMRSA-15 clone in a tertiary care Portuguese hospital. J Clin Microbiol 45: 2881–2888 - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases