

Powerful identification of cis-regulatory SNPs in human primary monocytes using allele-specific gene expression
- PMID: 23300628
- PMCID: PMC3530574
- DOI: 10.1371/journal.pone.0052260
Powerful identification of cis-regulatory SNPs in human primary monocytes using allele-specific gene expression
Abstract
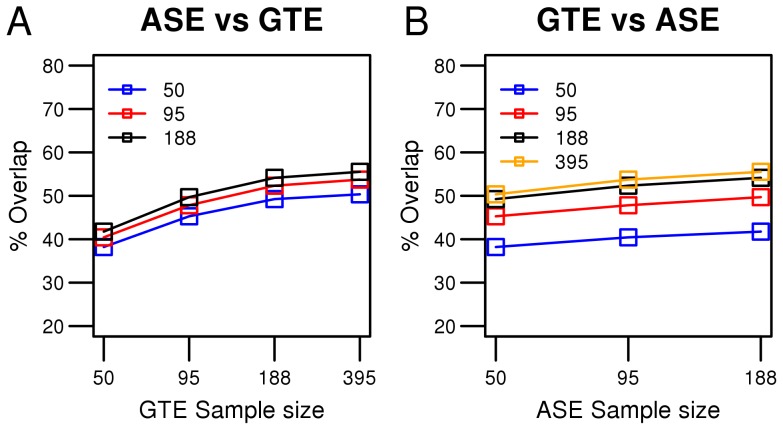
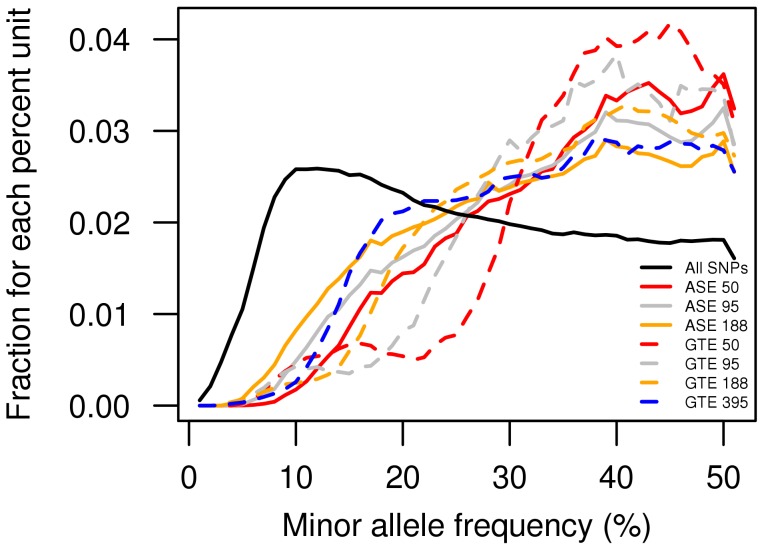
A large number of genome-wide association studies have been performed during the past five years to identify associations between SNPs and human complex diseases and traits. The assignment of a functional role for the identified disease-associated SNP is not straight-forward. Genome-wide expression quantitative trait locus (eQTL) analysis is frequently used as the initial step to define a function while allele-specific gene expression (ASE) analysis has not yet gained a wide-spread use in disease mapping studies. We compared the power to identify cis-acting regulatory SNPs (cis-rSNPs) by genome-wide allele-specific gene expression (ASE) analysis with that of traditional expression quantitative trait locus (eQTL) mapping. Our study included 395 healthy blood donors for whom global gene expression profiles in circulating monocytes were determined by Illumina BeadArrays. ASE was assessed in a subset of these monocytes from 188 donors by quantitative genotyping of mRNA using a genome-wide panel of SNP markers. The performance of the two methods for detecting cis-rSNPs was evaluated by comparing associations between SNP genotypes and gene expression levels in sample sets of varying size. We found that up to 8-fold more samples are required for eQTL mapping to reach the same statistical power as that obtained by ASE analysis for the same rSNPs. The performance of ASE is insensitive to SNPs with low minor allele frequencies and detects a larger number of significantly associated rSNPs using the same sample size as eQTL mapping. An unequivocal conclusion from our comparison is that ASE analysis is more sensitive for detecting cis-rSNPs than standard eQTL mapping. Our study shows the potential of ASE mapping in tissue samples and primary cells which are difficult to obtain in large numbers.
Conflict of interest statement
Figures
References
-
- Hindorff LA, MacArthur J, Wise A, Junkins HA, Hall PN, et al.. (2011) A Catalog of Published Genome-Wide Association Studies. Available: www.genome.gov/gwastudies. Accessed 2010 Oct 15.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
