

Claudin-19 mutations and clinical phenotype in Spanish patients with familial hypomagnesemia with hypercalciuria and nephrocalcinosis
- PMID: 23301036
- PMCID: PMC3536807
- DOI: 10.1371/journal.pone.0053151
Claudin-19 mutations and clinical phenotype in Spanish patients with familial hypomagnesemia with hypercalciuria and nephrocalcinosis
Abstract
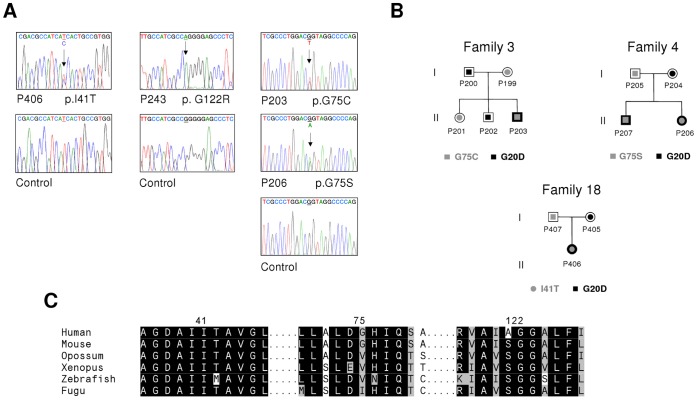
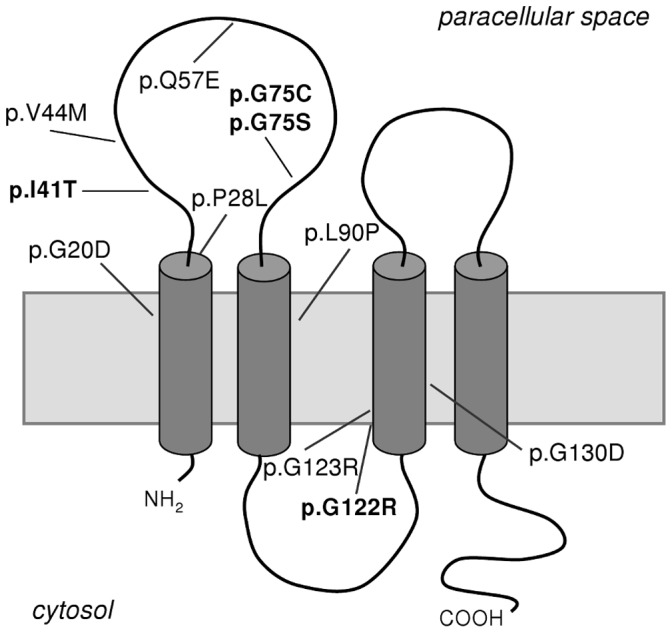
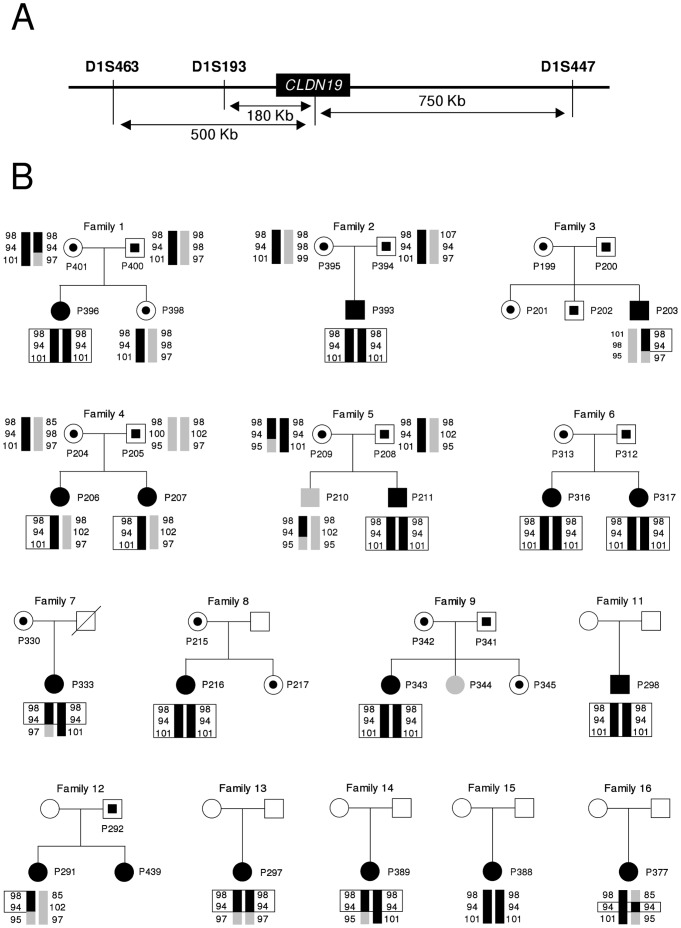
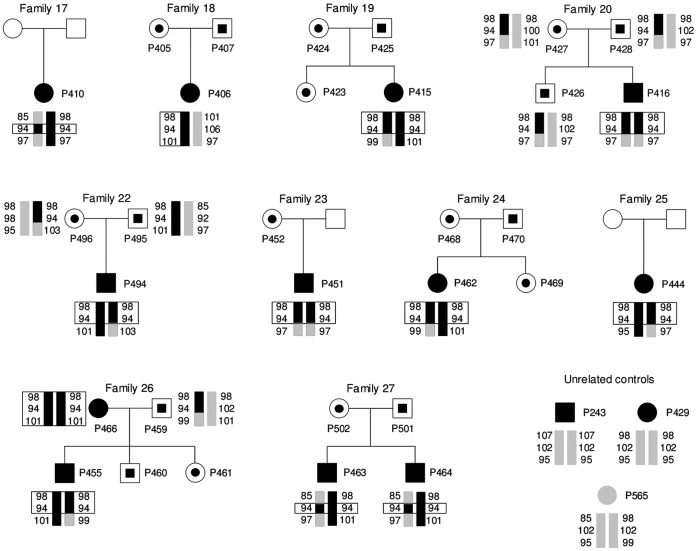
Familial hypomagnesemia with hypercalciuria and nephrocalcinosis is an autosomal recessive tubular disorder characterized by excessive renal magnesium and calcium excretion and chronic kidney failure. This rare disease is caused by mutations in the CLDN16 and CLDN19 genes. These genes encode the tight junction proteins claudin-16 and claudin-19, respectively, which regulate the paracellular ion reabsorption in the kidney. Patients with mutations in the CLDN19 gene also present severe visual impairment. Our goals in this study were to examine the clinical characteristics of a large cohort of Spanish patients with this disorder and to identify the disease causing mutations. We included a total of 31 patients belonging to 27 unrelated families and studied renal and ocular manifestations. We then analyzed by direct DNA sequencing the coding regions of CLDN16 and CLDN19 genes in these patients. Bioinformatic tools were used to predict the consequences of mutations. Clinical evaluation showed ocular defects in 87% of patients, including mainly myopia, nystagmus and macular colobomata. Twenty two percent of patients underwent renal transplantation and impaired renal function was observed in another 61% of patients. Results of the genetic analysis revealed CLDN19 mutations in all patients confirming the clinical diagnosis. The majority of patients exhibited the previously described p.G20D mutation. Haplotype analysis using three microsatellite markers showed a founder effect for this recurrent mutation in our cohort. We also identified four new pathogenic mutations in CLDN19, p.G122R, p.I41T, p.G75C and p.G75S. A strategy based on microsequencing was designed to facilitate the genetic diagnosis of this disease. Our data indicate that patients with CLDN19 mutations have a high risk of progression to chronic renal disease.
Conflict of interest statement
Figures
References
-
- Praga M, Vara J, Gonzalez-Parra E, Andres A, Alamo C, et al. (1995) Familial hypomagnesemia with hypercalciuria and nephrocalcinosis. Kidney Int 47: 1419–1425. - PubMed
-
- Rodríguez-Soriano J, Vallo A, García-Fuentes M (1987) Hypomagnesaemia of hereditary renal origin. Pediatr Nephrol 1: 465–472. - PubMed
-
- Weber S, Schneider L, Peters M, Misselwitz J, Ronnefarth G, et al. (2001) Novel paracellin-1 mutations in 25 families with familial hypomagnesemia with hypercalciuria and nephrocalcinosis. J Am Soc Nephrol 12: 1872–1881. - PubMed
-
- Benigno V, Canonica CS, Bettinelli A, von Vigier RO, Truttmann AC, et al. (2000) Hypomagnesaemia-hypercalciuria-nephrocalcinosis: A report of nine cases and a review. Nephrol Dial Transplant 15: 605–610. - PubMed
-
- Simon DB, Lu Y, Choate KA, Velazquez H, Al-Sabban E, et al. (1999) Paracellin-1, a renal tight junction protein required for paracellular Mg2+ resorption. Science 285: 103–106. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
