

Review
doi: 10.1021/ac303064a.
Epub 2013 Feb 19.
Integral membrane proteins and bilayer proteomics
Affiliations
- PMID: 23301778
- PMCID: PMC3664232
- DOI: 10.1021/ac303064a
Item in Clipboard
Review
Integral membrane proteins and bilayer proteomics
Anal Chem.
.
Abstract
Integral membrane proteins reside within the bilayer membranes that surround cells and organelles, playing critical roles in movement of molecules across them and the transduction of energy and signals. While their extreme amphipathicity presents technical challenges, biological mass spectrometry has been applied to all aspects of membrane protein chemistry and biology, including analysis of primary, secondary, tertiary, and quaternary structures as well as the dynamics that accompany functional cycles and catalysis.
Figures

Structural motifs of integral membrane proteins. Integral membrane proteins constitute a significant component of biological membranes and have domains that span the lipid bilayer. The most common motif is the transmembrane alpha-helix that is often multiplexed generating a polyhelix bundle such as that shown for bacteriorhodopsin (A). Since the alpha-helix is constructed from a continuous sequence, it is typical that 20–30 apolar amino-acid residues span the membrane, generating a hydrophobic stretch of primary structure – the red spirals that dominate the figure. The other common motif is the transmembrane beta-barrel, as illustrated with murine VDAC1 (B). The beta structure alternates hydrophobic side-chains toward the interior of the membrane and more polar side-chains toward the hydrated center of the pore. Disruption of the bilayer using detergents or organic solvents is necessary to extract whole integral membrane proteins for analysis while surface ‘shaving’ proteolysis experiments can be used to release peptides for mass spectrometry. The VDAC1 structure was reproduced with permission, Copyright (2008) National Academy of Sciences, USA.
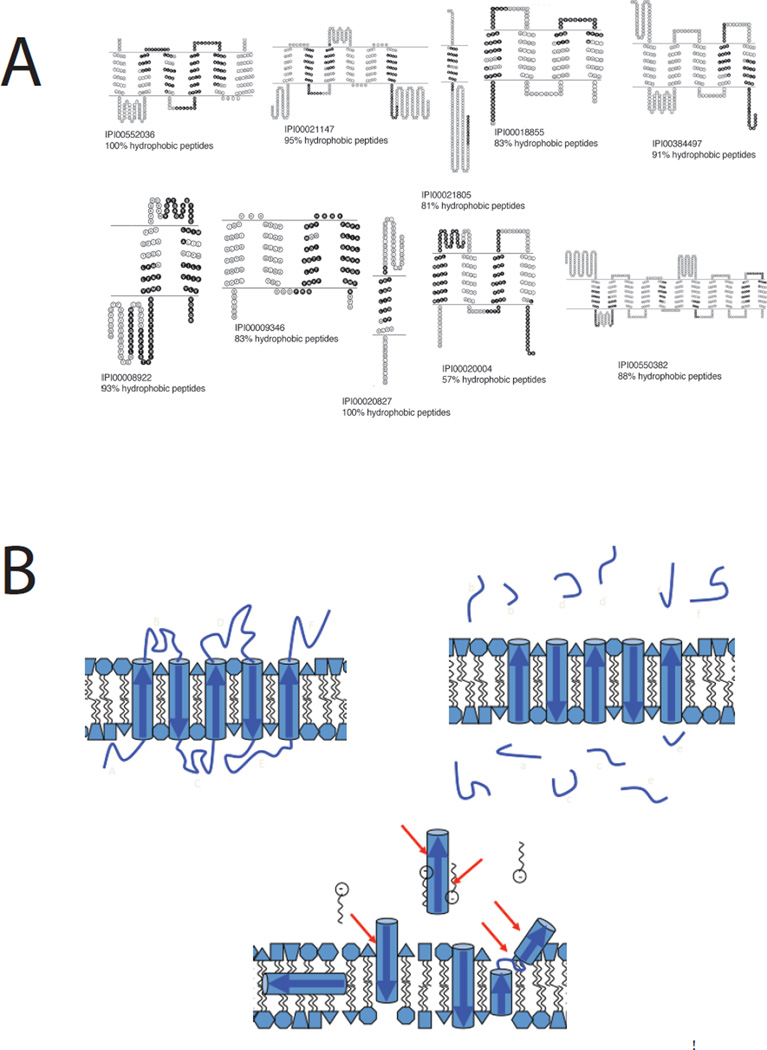
Peptide recovery in shotgun proteomics. Sophisticated protocols are dramatically improving representation of integral membrane proteins in shot-gun proteomics experiments. A. After shaving soluble loop regions with proteinase K at high pH to release protease-accessible peptides (PAPs) and subsequent cleavage of membrane-embedded polypeptides (MEPs) with Met-directed CNBr both fractions are analyzed by MuDPIT at elevated column temperature. The black shaded polypeptide regions recovered from the MEP fraction resulted in many protein identifications not seen in the PAP fraction (57% of total) and raised the proportion of integral membrane proteins recovered to 38% of the total, in line with the number predicted by informatics approaches. Note that despite landmark advances in proteolysis efficiency and peptide recovery, sequence coverage within the transmembrane domains remains sporadic. Reproduced from reference 27. B. A schematic shows how proteolytic shaving works under idealistic conditions, releasing loop regions for mass spectrometric analysis (upper right). The lower image represents what may happen during a lengthy proteolysis experiment as membrane protein structures react to early cleavage events, emphasizing that interpretation of such experiments should proceed with caution.
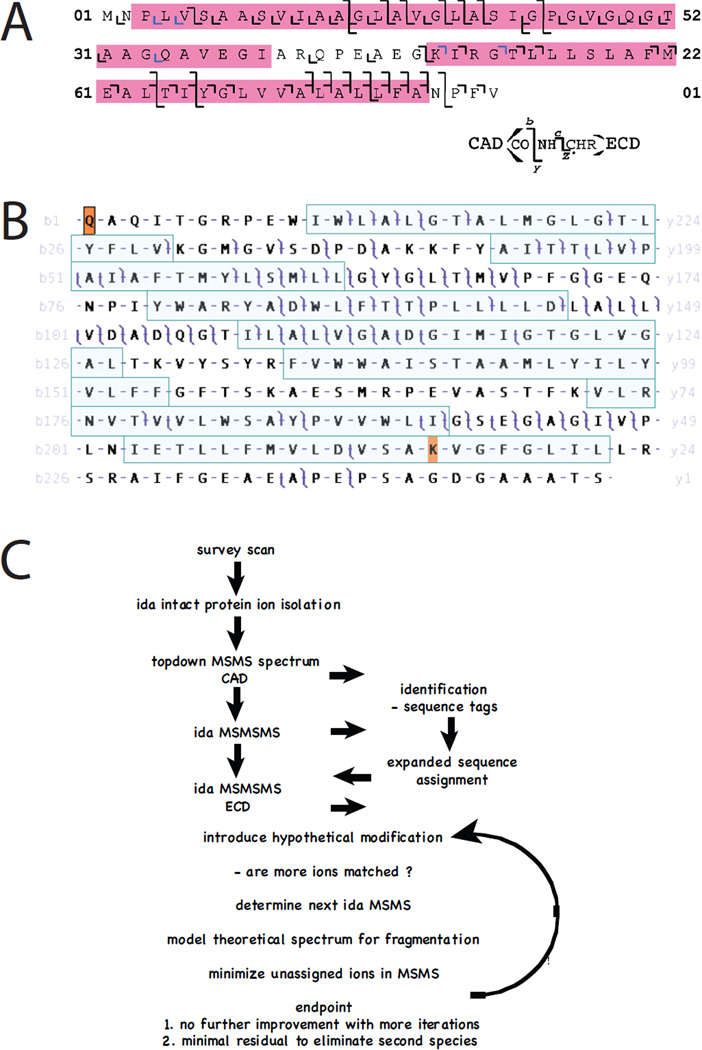
Top-down mass spectrometry of integral membrane proteins. A. Sequence coverage within the transmembrane domains of the c-subunit of chloroplast ATP synthase was improved when activated-ion electron-capture dissociation (aiECD) was compared with collisionally activcated dissociation (CAD). c and z product ions colored blue were assigned manually. Reproduced from reference 71. B. Multiply charged ions generated by electrospray-ionization of intact bacteriorhodopsin were subjected to collisionally activated dissociation (CAD) and product ions analyzed with a high-resolution FT-ICR mass spectrometer. Masses of precursor and product ions were matched at 10 ppm tolerance to the known sequence of the protein with post-translational removal of residues 1 – 13, N-terminal cyclization to form pyrrolidone carboxylic acid, removal of Asp262, and modification of Lys229 with N6-(retinylidene)lysine. Note that many product ions yield overlapping b- and y- ions such that the entire sequence is covered. While some transmembrane helices (shaded) are accessible to CAD, other regions are less so, with their primary structures inferred based upon the genomic translation and the numerous precursor and product ions that span them. Reproduced with permission from Mol. Cell. Proteomics 2010, 9, 791–803, copyright ASBMB. C. In the future data will be interpreted ‘on the fly’ so that experiments are controled in real time to improve coverage across primary sequence and post-translational modifications using MS strategies.
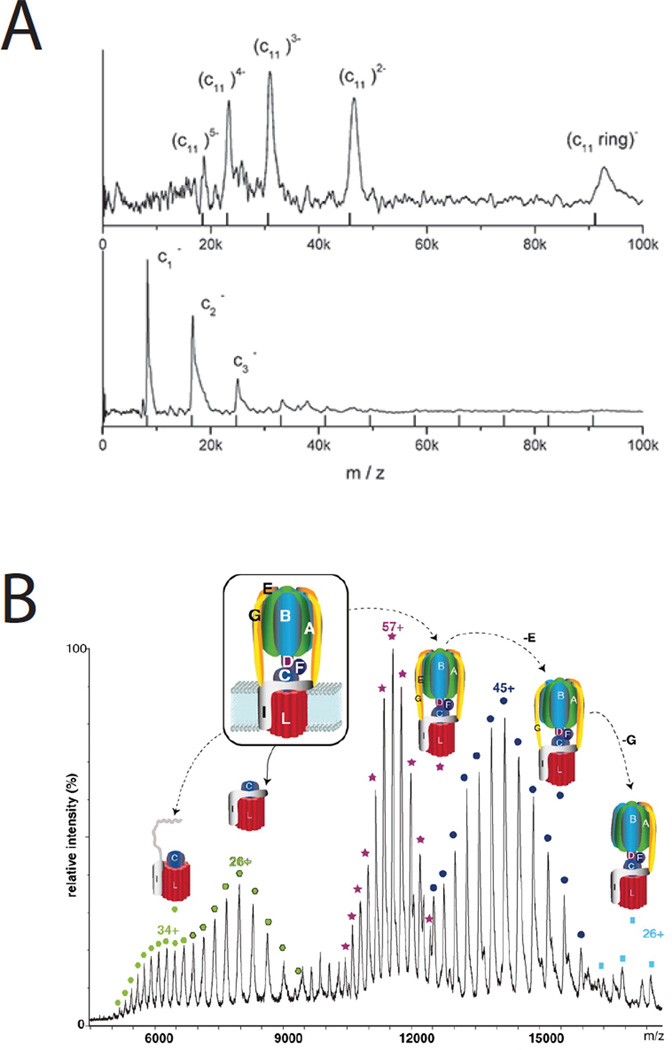
Native mass spectrometry of integral membrane protein complexes. There has been long standing interest in the number of c-subunits that make up the ring component of Fo of the ATP synthase. A. The soft ionization technique laser-induced liquid bead ion desorption-MS (LILBID) was used to directly measure c-ring stoichiometry in a number of prokaryotic complexes including Clostridium paradoxum with 11 subunits - the upper panel used minimal laser excitation to retain the intact native complex, while the lower panel uses higher excitation shifting the spectrum toward the monomer. The mass spectra were recorded using a time-of-flight analyzer operated in negative ion mode. Reproduced with permission from reference 83. B. Positive-ion electrospray-ionization was used to measure the m/z of intact ATP-synthase complexes, and sub-complexes where various subunits had been displaced in the gas phase. The complex shown is from Enterococcus hirae and has a measured c-ring stoichiometry of 12. Six molecules of the phospholipid phosphatidylethanolamine were bound to each c-ring. Reproduced from reference 86. Printed with permission from AAAS.
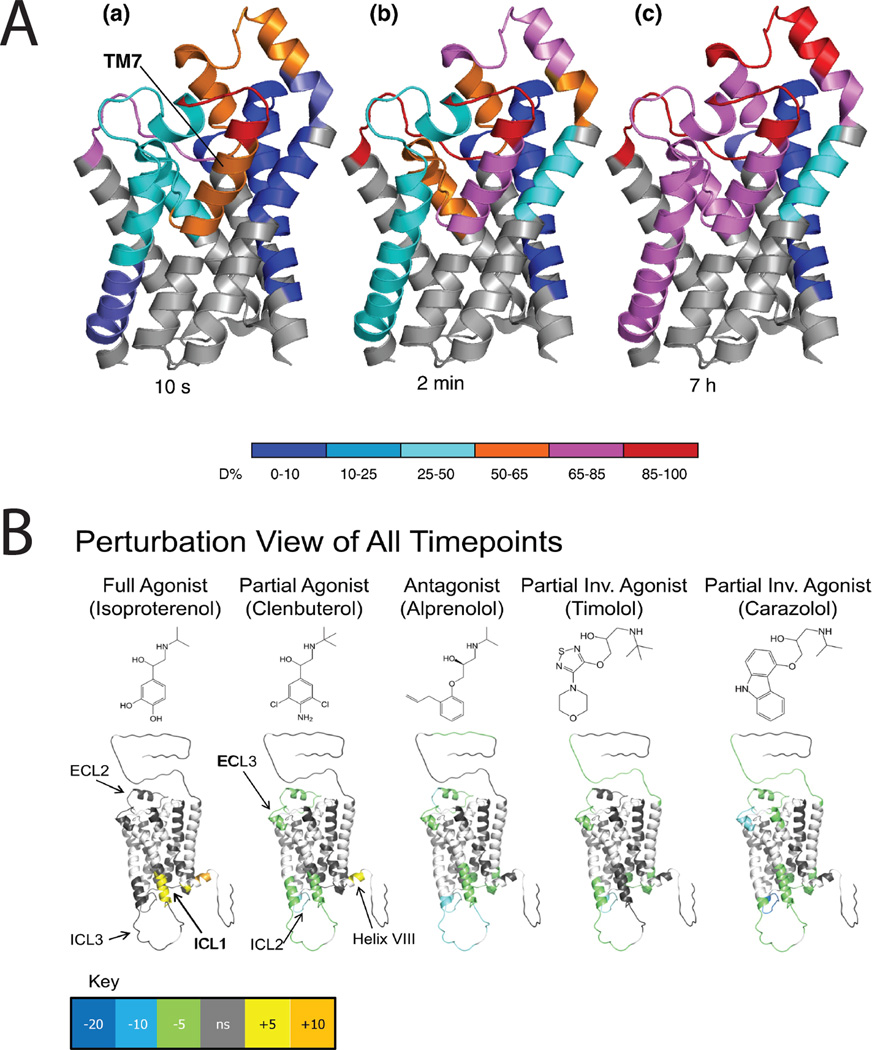
Hydrogen-deuterium exchange (HDX) provides insights into dynamics of integral membrane proteins. A. Transmembrane helix 7 of the glycerol facilitator (GF) is highly dynamic. Deuteration behavior of GF depicted by color-coding individual protein segments according to their deuteration levels. (a) t = 10 s. (b) t = 2 min. (c) t = 7 h. Note that exchange requires breakage of alpha-helix backbone amide H-bonds. Reprinted from J. Mol. Biol. 416(3). Pan Y, Piyadasa H, O'Neil JD, Konermann L. Conformational dynamics of a membrane transport protein probed by H/D exchange and covalent labeling: the glycerol facilitator. 400–413. Copyright 2012, with permission from Elsevier. B. HDX data overlaid onto a modified crystal structure for the beta-adrenergic receptor (β2AR). The HDX data are shown using color gradients where the differences in the average percentage deuterium uptake across all time points between the apo- and ligand-bound receptor for peptides in a given region of the receptor sequence are assigned a color as shown in the key, and then overlaid onto the structure. Here, white indicates regions of the receptor that were not resolved in the HDX experiment, and gray indicates no significant change. Ligand structures are indicated above the protein structures. Reproduced from Structure 19(10), West GM, Chien EY, Katritch V, Gatchalian J, Chalmers MJ, Stevens RC, Griffin PR. Ligand-dependent perturbation of the conformational ensemble for the GPCR β2 adrenergic receptor revealed by HDX. 1424-32. Copyright 2011, with permission from Elsevier.
References
-
- Fagerberg L, Jonasson K, von Heijne G, Uhlén M, Berglund L. Proteomics. 2010;10(6):1141–1149. - PubMed
-
- Weinglass AB, Whitelegge JP, Kaback HR. Curr. Opin. Drug Discov. Devel. 2004;7(5):589–599. - PubMed
-
- von Heijne G. Annu. Rev. Biochem. 2011;80:157–160. - PubMed
-
- von Heijne G. Biochem. Soc. Trans. 2011;39(3):747–750. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
