

Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance
- PMID: 23302800
- PMCID: PMC3930354
- DOI: 10.1038/nature11814
Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance
Abstract
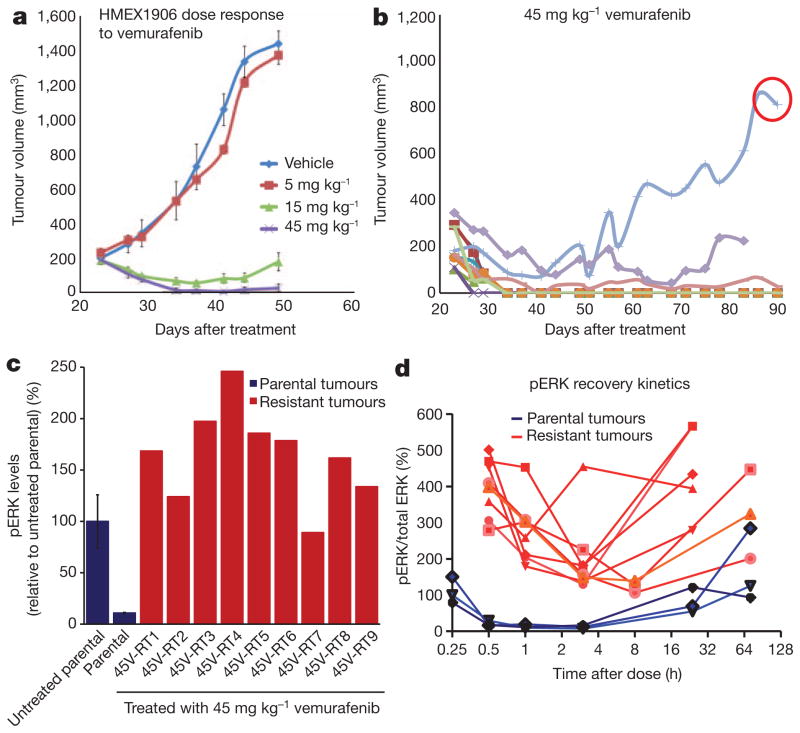
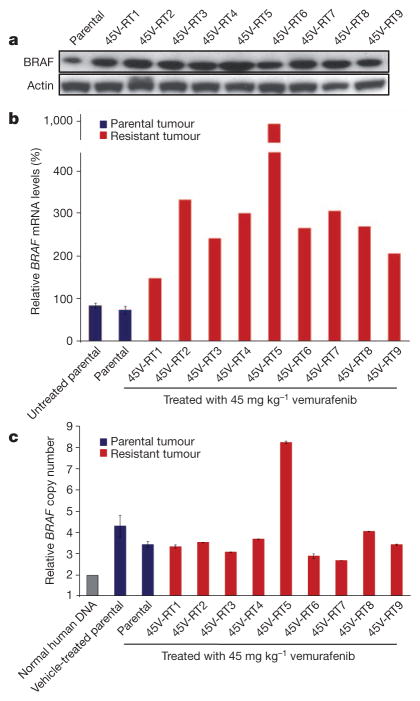
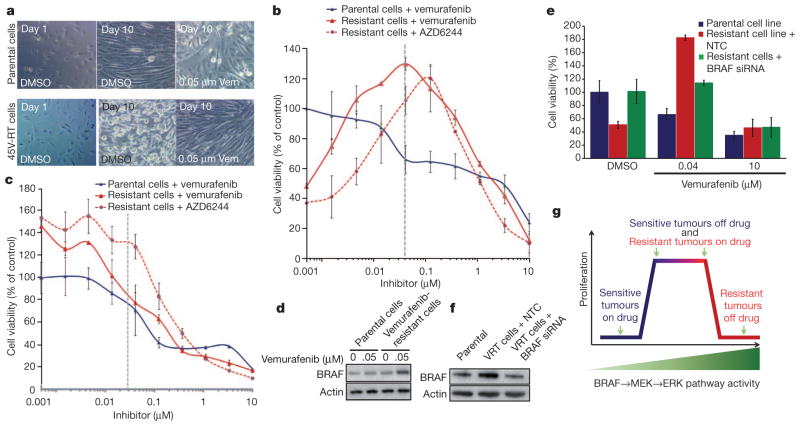
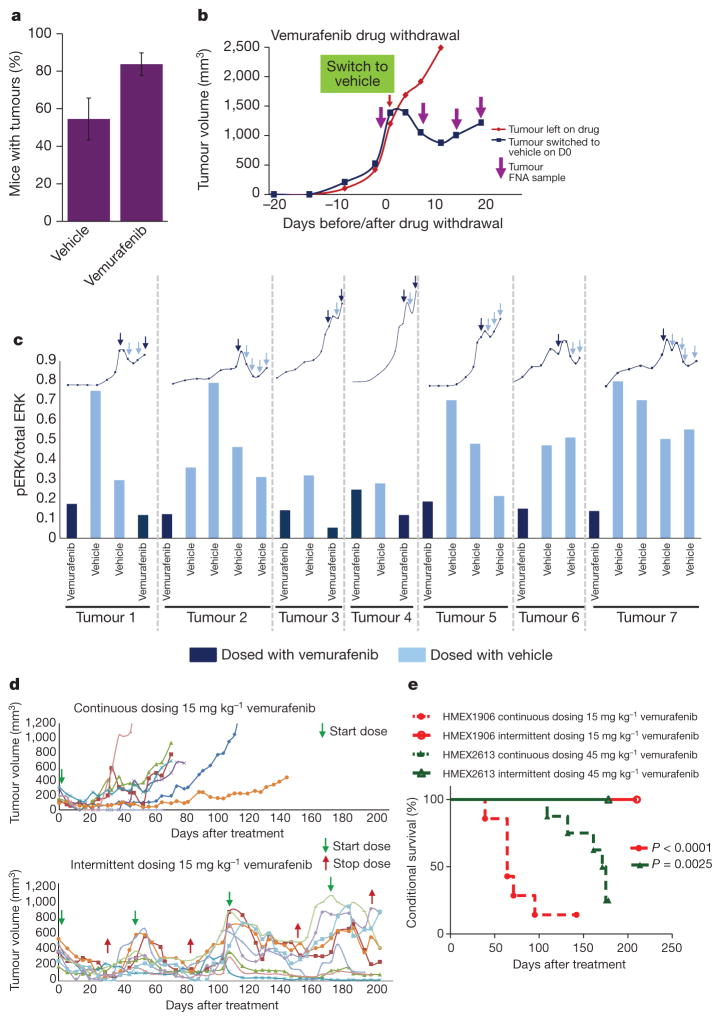
Mutational activation of BRAF is the most prevalent genetic alteration in human melanoma, with ≥50% of tumours expressing the BRAF(V600E) oncoprotein. Moreover, the marked tumour regression and improved survival of late-stage BRAF-mutated melanoma patients in response to treatment with vemurafenib demonstrates the essential role of oncogenic BRAF in melanoma maintenance. However, as most patients relapse with lethal drug-resistant disease, understanding and preventing mechanism(s) of resistance is critical to providing improved therapy. Here we investigate the cause and consequences of vemurafenib resistance using two independently derived primary human melanoma xenograft models in which drug resistance is selected by continuous vemurafenib administration. In one of these models, resistant tumours show continued dependency on BRAF(V600E)→MEK→ERK signalling owing to elevated BRAF(V600E) expression. Most importantly, we demonstrate that vemurafenib-resistant melanomas become drug dependent for their continued proliferation, such that cessation of drug administration leads to regression of established drug-resistant tumours. We further demonstrate that a discontinuous dosing strategy, which exploits the fitness disadvantage displayed by drug-resistant cells in the absence of the drug, forestalls the onset of lethal drug-resistant disease. These data highlight the concept that drug-resistant cells may also display drug dependency, such that altered dosing may prevent the emergence of lethal drug resistance. Such observations may contribute to sustaining the durability of the vemurafenib response with the ultimate goal of curative therapy for the subset of melanoma patients with BRAF mutations.
Figures
Comment in
-
Skin cancer: Novel mouse model reveals strategy to delay drug resistance in melanoma.Nat Rev Clin Oncol. 2013 Mar;10(3):123. doi: 10.1038/nrclinonc.2013.11. Epub 2013 Jan 22. Nat Rev Clin Oncol. 2013. PMID: 23337922 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
