

A novel approach, based on BLSOMs (Batch Learning Self-Organizing Maps), to the microbiome analysis of ticks
- PMID: 23303373
- PMCID: PMC3635243
- DOI: 10.1038/ismej.2012.171
A novel approach, based on BLSOMs (Batch Learning Self-Organizing Maps), to the microbiome analysis of ticks
Erratum in
- ISME J. 2014 Aug;8(8):1752
Abstract
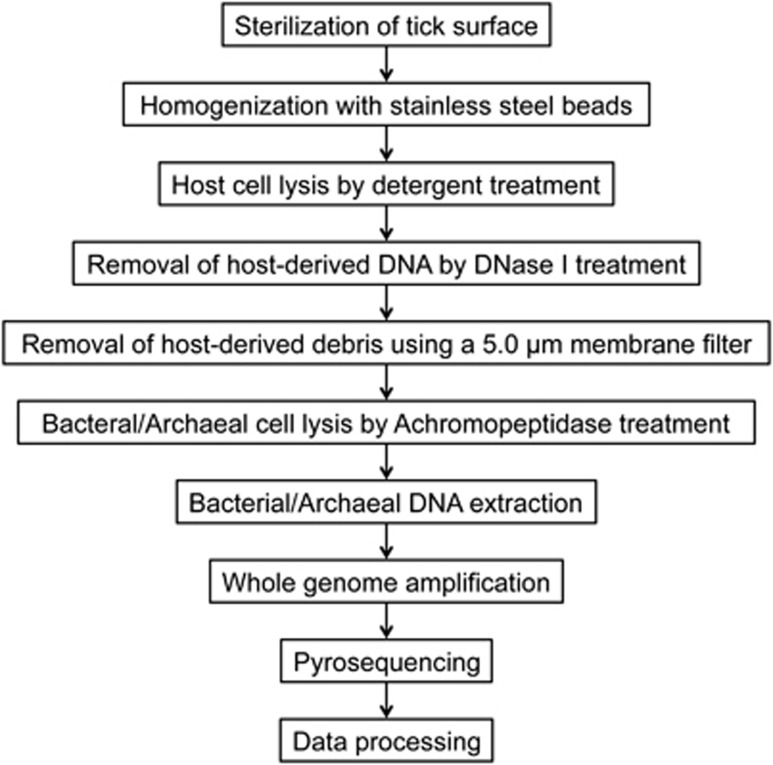
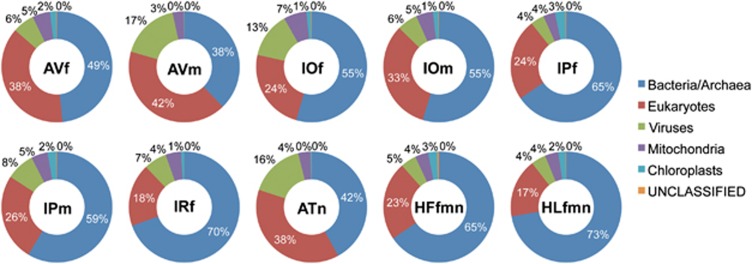
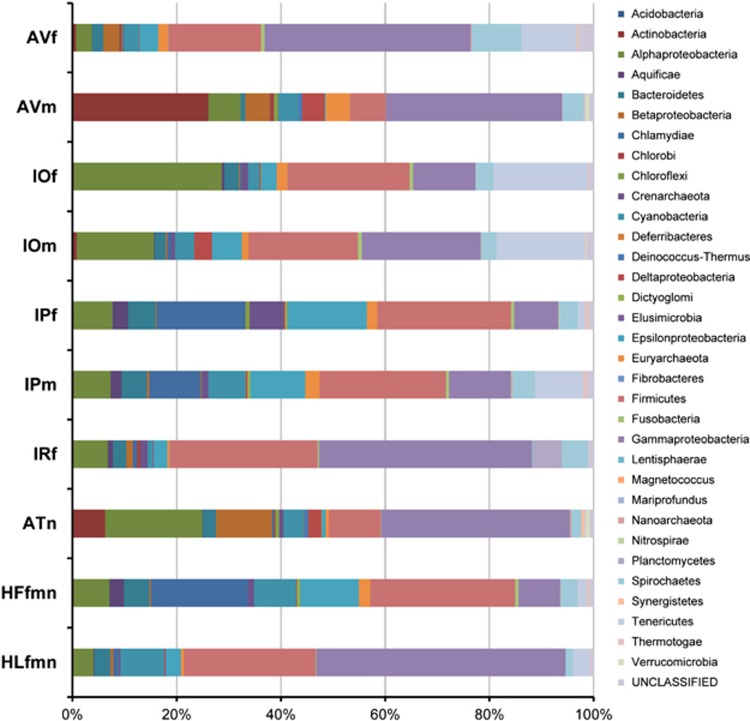
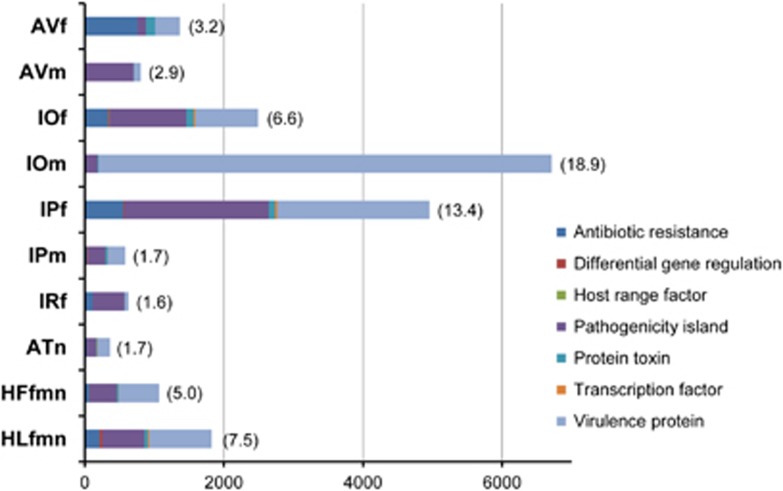
Ticks transmit a variety of viral, bacterial and protozoal pathogens, which are often zoonotic. The aim of this study was to identify diverse tick microbiomes, which may contain as-yet unidentified pathogens, using a metagenomic approach. DNA prepared from bacteria/archaea-enriched fractions obtained from seven tick species, namely Amblyomma testudinarium, Amblyomma variegatum, Haemaphysalis formosensis, Haemaphysalis longicornis, Ixodes ovatus, Ixodes persulcatus and Ixodes ricinus, was subjected to pyrosequencing after whole-genome amplification. The resulting sequence reads were phylotyped using a Batch Learning Self-Organizing Map (BLSOM) program, which allowed phylogenetic estimation based on similarity of oligonucleotide frequencies, and functional annotation by BLASTX similarity searches. In addition to bacteria previously associated with human/animal diseases, such as Anaplasma, Bartonella, Borrelia, Ehrlichia, Francisella and Rickettsia, BLSOM analysis detected microorganisms belonging to the phylum Chlamydiae in some tick species. This was confirmed by pan-Chlamydia PCR and sequencing analysis. Gene sequences associated with bacterial pathogenesis were also identified, some of which were suspected to originate from horizontal gene transfer. These efforts to construct a database of tick microbes may lead to the ability to predict emerging tick-borne diseases. Furthermore, a comprehensive understanding of tick microbiomes will be useful for understanding tick biology, including vector competency and interactions with pathogens and symbionts.
Figures
Similar articles
-
Viral population analysis of the taiga tick, Ixodes persulcatus, by using Batch Learning Self-Organizing Maps and BLAST search.J Vet Med Sci. 2019 Mar 20;81(3):401-410. doi: 10.1292/jvms.18-0483. Epub 2019 Jan 23. J Vet Med Sci. 2019. PMID: 30674747 Free PMC article.
-
Detection of tick-borne pathogens in blood-fed ticks from animals across nine Asian countries.Microbiol Spectr. 2025 Mar 4;13(3):e0244924. doi: 10.1128/spectrum.02449-24. Epub 2025 Feb 4. Microbiol Spectr. 2025. PMID: 39902978 Free PMC article.
-
Co-Infection Patterns in Individual Ixodes scapularis Ticks Reveal Associations between Viral, Eukaryotic and Bacterial Microorganisms.Viruses. 2018 Jul 22;10(7):388. doi: 10.3390/v10070388. Viruses. 2018. PMID: 30037148 Free PMC article.
-
[Ticks (Acarina: Ixodidae) as vectors and reservoirs of pathogen microorganisms in Spain].Enferm Infecc Microbiol Clin. 2005 Feb;23(2):94-102. doi: 10.1157/13071613. Enferm Infecc Microbiol Clin. 2005. PMID: 15743581 Review. Spanish.
-
The Tick Microbiome: Why Non-pathogenic Microorganisms Matter in Tick Biology and Pathogen Transmission.Front Cell Infect Microbiol. 2017 Jun 8;7:236. doi: 10.3389/fcimb.2017.00236. eCollection 2017. Front Cell Infect Microbiol. 2017. PMID: 28642842 Free PMC article. Review.
Cited by
-
Tick Microbiomes in Neotropical Forest Fragments Are Best Explained by Tick-Associated and Environmental Factors Rather than Host Blood Source.Appl Environ Microbiol. 2021 Mar 11;87(7):e02668-20. doi: 10.1128/AEM.02668-20. Print 2021 Mar 11. Appl Environ Microbiol. 2021. PMID: 33514519 Free PMC article.
-
Cross-alteration of murine skin and tick microbiome concomitant with pathogen transmission after Ixodes ricinus bite.Microbiome. 2023 Nov 11;11(1):250. doi: 10.1186/s40168-023-01696-7. Microbiome. 2023. PMID: 37952001 Free PMC article.
-
Comparative Metagenomic Profiling of Symbiotic Bacterial Communities Associated with Ixodes persulcatus, Ixodes pavlovskyi and Dermacentor reticulatus Ticks.PLoS One. 2015 Jul 8;10(7):e0131413. doi: 10.1371/journal.pone.0131413. eCollection 2015. PLoS One. 2015. PMID: 26154300 Free PMC article.
-
Ticks and Chlamydia-Related Bacteria in Swiss Zoological Gardens Compared to in Contiguous and Distant Control Areas.Microorganisms. 2023 Sep 30;11(10):2468. doi: 10.3390/microorganisms11102468. Microorganisms. 2023. PMID: 37894126 Free PMC article.
-
A Roadmap for Tick-Borne Flavivirus Research in the "Omics" Era.Front Cell Infect Microbiol. 2017 Dec 22;7:519. doi: 10.3389/fcimb.2017.00519. eCollection 2017. Front Cell Infect Microbiol. 2017. PMID: 29312896 Free PMC article. Review.
References
-
- Abe T, Sugawara H, Kinouchi M, Kanaya S, Ikemura T. Novel phylogenetic studies of genomic sequence fragments derived from uncultured microbe mixtures in environmental and clinical samples. DNA Res. 2005;12:281–290. - PubMed
-
- Allen HK, Donato J, Wang HH, Cloud-Hansen KA, Davies J, Handelsman J. Call of the wild: antibiotic resistance genes in natural environments. Nat Rev Microbiol. 2010;8:251–259. - PubMed
-
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
