

Extending the coverage of spectral libraries: a neighbor-based approach to predicting intensities of peptide fragmentation spectra
- PMID: 23303707
- PMCID: PMC3733334
- DOI: 10.1002/pmic.201100670
Extending the coverage of spectral libraries: a neighbor-based approach to predicting intensities of peptide fragmentation spectra
Abstract
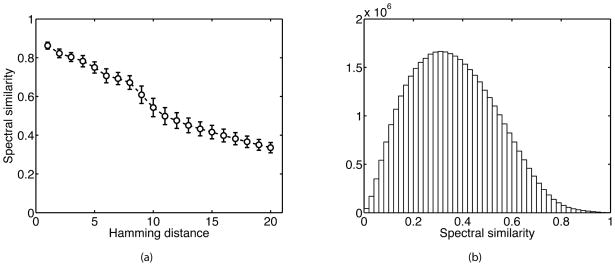
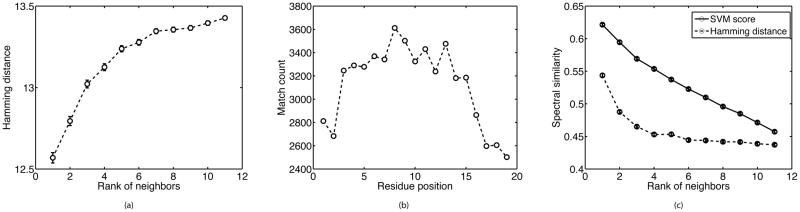
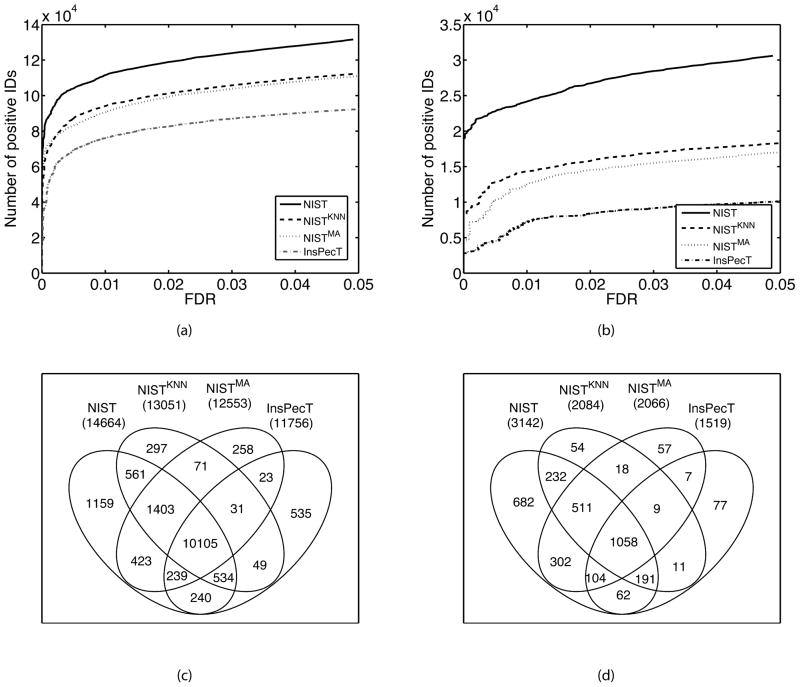
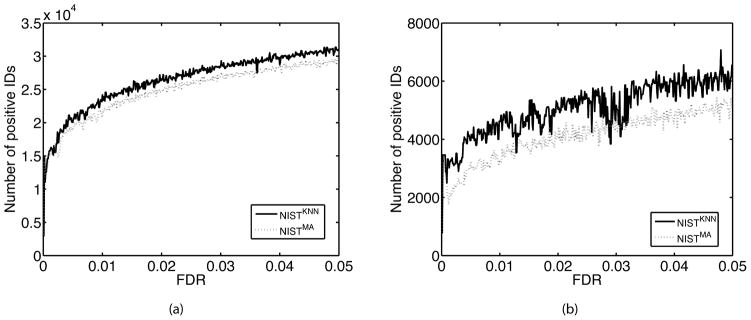
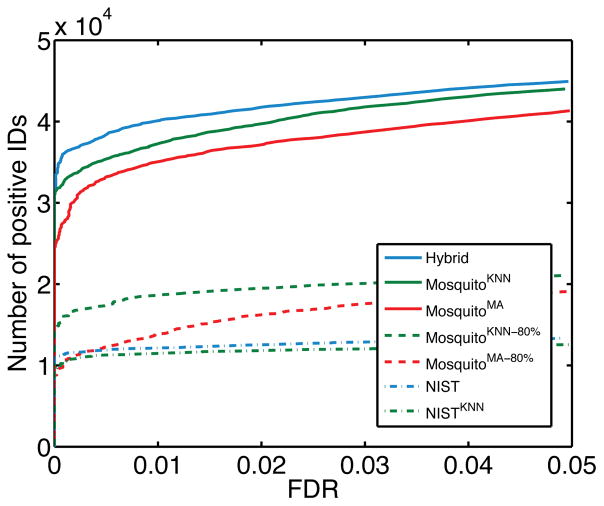
Searching spectral libraries in MS/MS is an important new approach to improving the quality of peptide and protein identification. The idea relies on the observation that ion intensities in an MS/MS spectrum of a given peptide are generally reproducible across experiments, and thus, matching between spectra from an experiment and the spectra of previously identified peptides stored in a spectral library can lead to better peptide identification compared to the traditional database search. However, the use of libraries is greatly limited by their coverage of peptide sequences: even for well-studied organisms a large fraction of peptides have not been previously identified. To address this issue, we propose to expand spectral libraries by predicting the MS/MS spectra of peptides based on the spectra of peptides with similar sequences. We first demonstrate that the intensity patterns of dominant fragment ions between similar peptides tend to be similar. In accordance with this observation, we develop a neighbor-based approach that first selects peptides that are likely to have spectra similar to the target peptide and then combines their spectra using a weighted K-nearest neighbor method to accurately predict fragment ion intensities corresponding to the target peptide. This approach has the potential to predict spectra for every peptide in the proteome. When rigorous quality criteria are applied, we estimate that the method increases the coverage of spectral libraries available from the National Institute of Standards and Technology by 20-60%, although the values vary with peptide length and charge state. We find that the overall best search performance is achieved when spectral libraries are supplemented by the high quality predicted spectra.
© 2013 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.
Figures
Similar articles
-
A semi-empirical approach for predicting unobserved peptide MS/MS spectra from spectral libraries.Proteomics. 2011 Dec;11(24):4702-11. doi: 10.1002/pmic.201100316. Epub 2011 Nov 23. Proteomics. 2011. PMID: 22038894
-
Building and searching tandem mass (MS/MS) spectral libraries for peptide identification in proteomics.Methods. 2011 Aug;54(4):424-31. doi: 10.1016/j.ymeth.2011.01.007. Epub 2011 Jan 28. Methods. 2011. PMID: 21277371 Review.
-
Spectral Library Search Improves Assignment of TMT Labeled MS/MS Spectra.J Proteome Res. 2018 Sep 7;17(9):3325-3331. doi: 10.1021/acs.jproteome.8b00594. Epub 2018 Aug 16. J Proteome Res. 2018. PMID: 30096983 Free PMC article.
-
High-throughput database search and large-scale negative polarity liquid chromatography-tandem mass spectrometry with ultraviolet photodissociation for complex proteomic samples.Mol Cell Proteomics. 2013 Sep;12(9):2604-14. doi: 10.1074/mcp.O113.028258. Epub 2013 May 21. Mol Cell Proteomics. 2013. PMID: 23695934 Free PMC article.
-
Spectral library searching in proteomics.Proteomics. 2016 Mar;16(5):729-40. doi: 10.1002/pmic.201500296. Epub 2016 Feb 9. Proteomics. 2016. PMID: 26616598 Review.
Cited by
-
Impact of Amidination on Peptide Fragmentation and Identification in Shotgun Proteomics.J Proteome Res. 2016 Oct 7;15(10):3656-3665. doi: 10.1021/acs.jproteome.6b00468. Epub 2016 Sep 27. J Proteome Res. 2016. PMID: 27615690 Free PMC article.
-
MS2CNN: predicting MS/MS spectrum based on protein sequence using deep convolutional neural networks.BMC Genomics. 2019 Dec 24;20(Suppl 9):906. doi: 10.1186/s12864-019-6297-6. BMC Genomics. 2019. PMID: 31874640 Free PMC article.
-
Quantitative Comparison of Tandem Mass Spectra Obtained on Various Instruments.J Am Soc Mass Spectrom. 2016 Aug;27(8):1357-65. doi: 10.1007/s13361-016-1408-y. Epub 2016 May 20. J Am Soc Mass Spectrom. 2016. PMID: 27206510
References
-
- Aebersold R, Mann M. Mass spectrometry-based proteomics. Nature. 2003;422(6928):198–207. - PubMed
-
- Cravatt BF, Simon GM, Yates JR., 3rd The biological impact of mass-spectrometry-based proteomics. Nature. 2007;450(7172):991–1000. - PubMed
-
- Resing KA, Meyer-Arendt K, Mendoza AM, Aveline-Wolf LD, Jonscher KR, Pierce KG, Old WM, Cheung HT, Russell S, Wattawa JL, et al. Improving reproducibility and sensitivity in identifying human proteins by shotgun proteomics. Anal Chem. 2004;76(13):3556–3568. - PubMed
-
- Yen CY, Russell S, Mendoza AM, Meyer-Arendt K, Sun S, Cios KJ, Ahn NG, Resing KA. Improving sensitivity in shotgun proteomics using a peptide-centric database with reduced complexity: protease cleavage and SCX elution rules from data mining of MS/MS spectra. Anal Chem. 2006;78(4):1071–1084. - PubMed
-
- Resing KA, Ahn NG. Proteomics strategies for protein identification. FEBS Lett. 2005;579(4):885–889. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
