

Review
doi: 10.1101/gad.200014.112.
Regulation of AID, the B-cell genome mutator
Affiliations
- PMID: 23307864
- PMCID: PMC3553278
- DOI: 10.1101/gad.200014.112
Item in Clipboard
Review
Regulation of AID, the B-cell genome mutator
Genes Dev.
.
Abstract
The mechanisms by which B cells somatically engineer their genomes to generate the vast diversity of antibodies required to challenge the nearly infinite number of antigens that immune systems encounter are of tremendous clinical and academic interest. The DNA cytidine deaminase activation-induced deaminase (AID) catalyzes two of these mechanisms: class switch recombination (CSR) and somatic hypermutation (SHM). Recent discoveries indicate a significant promiscuous targeting of this B-cell mutator enzyme genome-wide. Here we discuss the various regulatory elements that control AID activity and prevent AID from inducing genomic instability and thereby initiating oncogenesis.
Figures
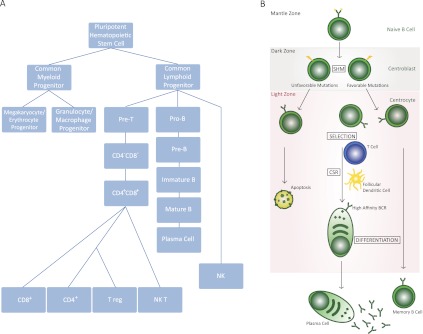
Development of B cells. (A) Development of various components of the immune system from HSCs. B cells emerge from the common lymphoid progenitor, a cell type that can differentiate to precursors of B or T cells. Pro-B cells continue their development to pre-B cells, which can then convert to mature B cells, often being converted to plasma cells in a GC-driven reaction. (B) A schematic of GC-based pathways that control B-cell development. The schematic highlights the various compartments of the GC where B cells undergo CSR and SHM following an encounter with an antigen. In addition, this schematic demonstrates that B cells are activated for CSR following interaction with T cells and follicular dendritic cells. The maturation of B cells due to SHM and CSR results from migration between the LZ and DZ of the GC, as discussed in the text.
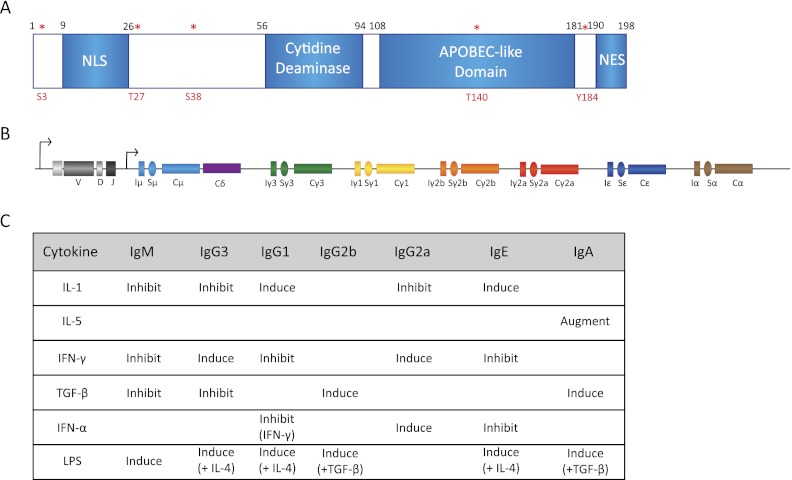
Essential components of CSR machinery. (A) Schematic representation of AID subdomains and various identified phosphorylation sites (in red asterisks). Note especially that (1) mutagenic analysis of AID protein has lead to identification of a nuclear localization signal (NLS) and a nuclear export signal (NES), (2) AID has a conserved cytidine deaminase motif, and (3) the phosphorylation sites S3, T27, S38, T140, and Y184 are characterized to a certain extent but require further work for complete functional characterization. (B) Structure of the Ig locus following VDJ recombination. Each constant region exon (Cx) is preceded by a cognate C sequence (Sx). The S sequences have their own transcriptional control elements that are activated by specific signaling pathways. (C) The combinations of various known cytokines that can activate S sequence transcription are shown. These cytokines often can be used to stimulate CSR in B cells grown in ex vivo cultures.
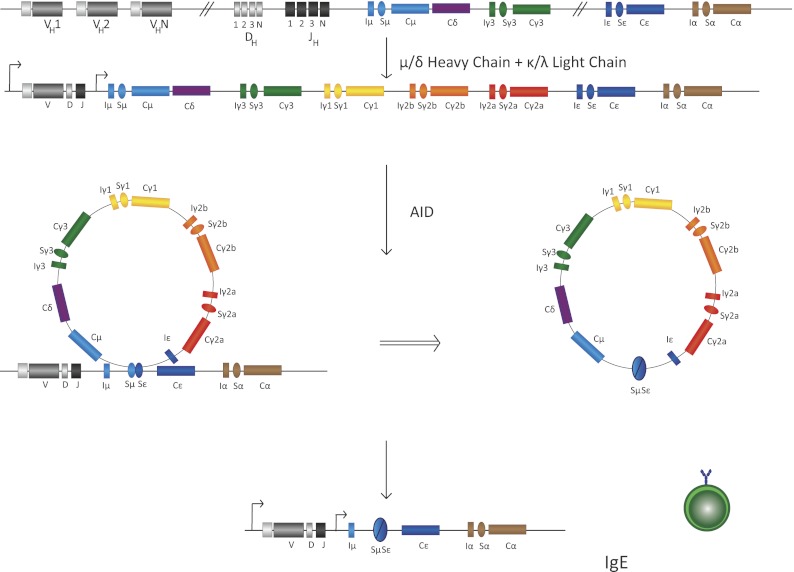
Schematic of CSR intermediate steps in B cells. Transcription of various S sequences, in this case IgSμ and IgSɛ, leads to generation of secondary DNA structures that are substrates of the ssDNA cytidine deaminase AID. AID-mediated DNA deamination leads to generation of DNA DSBs that are intermediates in a DNA deletion–recombination reaction, leading to completion of CSR. Recombination between S sequences leads to deletion of intermediate DNA sequences that loop out as a circle. In this case, the IgSμ recombines with the downstream IgSɛ, with the intermediate sequence shown to be looped out, leading to IgE CSR. B cells with this configuration of their IgH locus will be able to synthesize IgE. The details of each step in this illustration are discussed in the text.
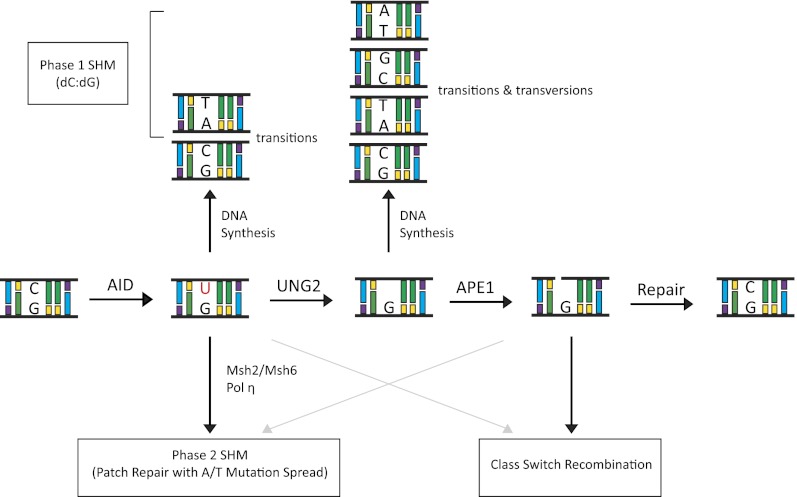
The DNA deamination model for AID activity in B cells. AID deaminates cytidine residues to uracils that are identified by the cellular BER pathway (UNG) or the MMR pathway (MSH2/MSH6) for repair. In this process of DNA lesion repair that depends on the DNA polymerase η, neighboring residues (A/T-based pairs) may be mutated. There are multiple possibilities how a lesion could be repaired. Based on the activity of error-prone DNA polymerases (DNA pol η), change of the neighboring A/T-based pair could be to T/A, G/C, or C/G base pairs. The nascently formed uracil residue can also be excised by the APEs (APE1/2) that lead to creation of ssDNA nicks. ssDNA nicks on both strands of S sequences can generate DNA DSBs that are intermediates during CSR.
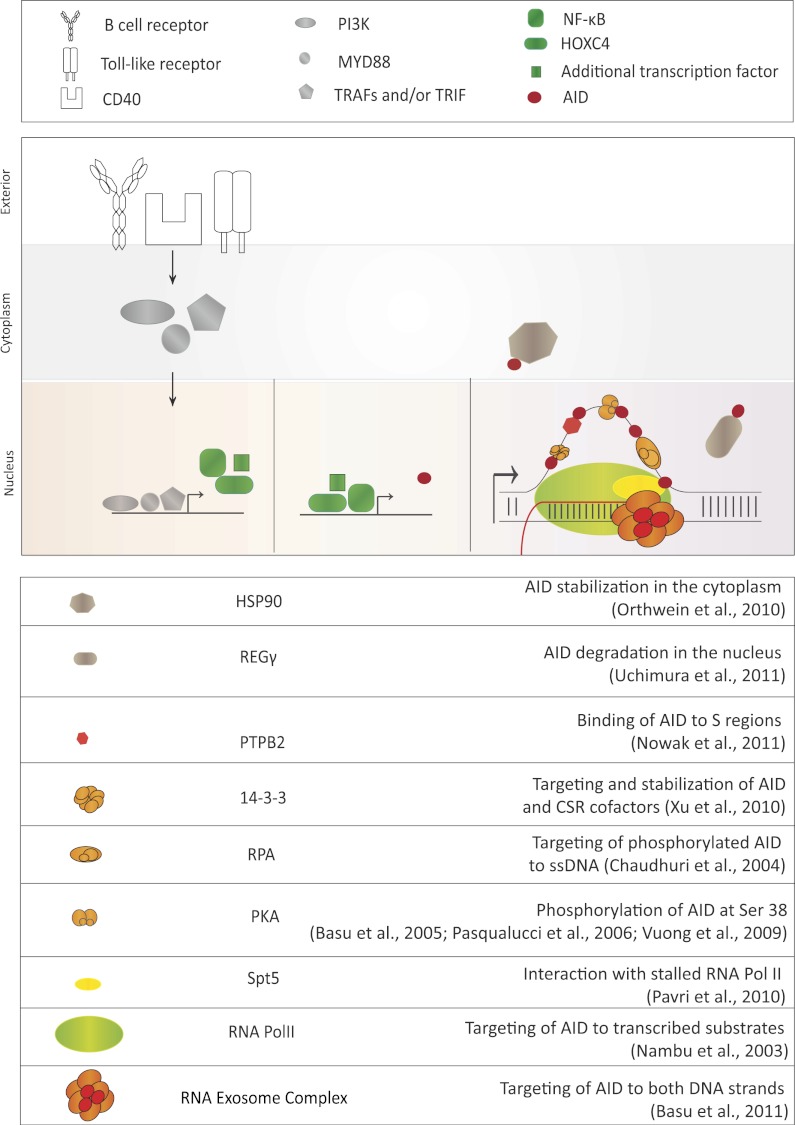
Various steps of AID regulation. AID expression is regulated by the transcription activation of the AID gene via various pathways. Activation of the B-cell receptor, CD40 receptor, or Toll-like receptor pathways stimulates AID locus transcription. Once AID is expressed, its activity is further regulated post-translationally by various identified AID cofactors listed in the table at the bottom of the figure. The proposed function of each identified cofactor is described, although further research will reveal the mechanism of AID cofactor complex function with additional detail. These factors can control AID's stability in the cytoplasm, regulate its nucleocytoplasmic distribution, stabilize its binding to target DNA sequences, and/or activate its DNA deamination activity.
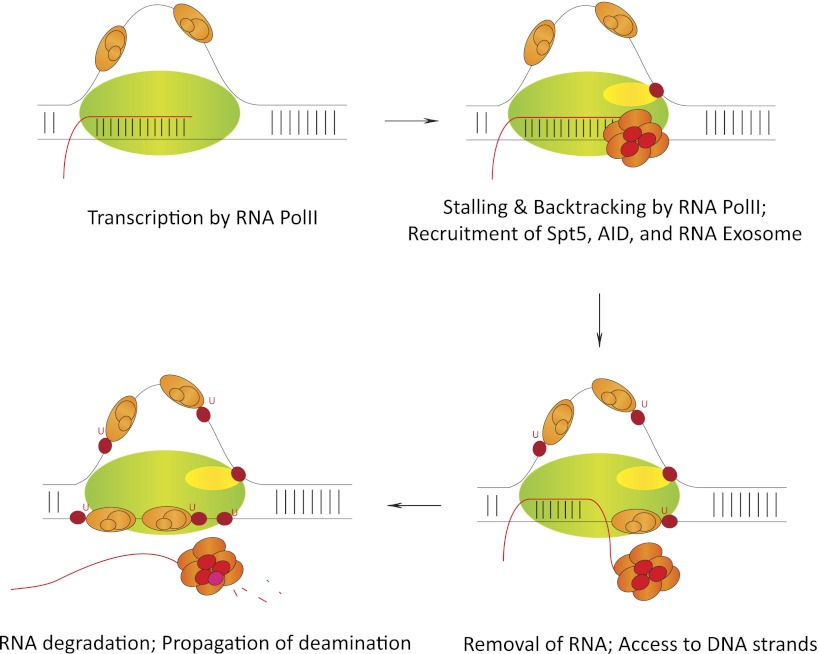
Step-by-step mechanism of cotranscriptional AID regulation. RNA polII, while transcribing AID's substrate sequences, stalls at various regions due to the formation of secondary DNA structures like R loops. Following stalling, AID (red) interacts with its cofactor, Spt5 (yellow). The ssDNA is stabilized by the ssDNA-binding protein complex RPA (orange). RNA exosome (multisubunit complex; red) binds the nascent transcript and excludes it from the DNA/RNA hybrid to present AID with both the template and nontemplate strand as DNA deamination substrates. In vivo, AID has been observed to mutate both template and nontemplate strands of transcribed S sequences. Details of this proposed AID deamination mechanism are discussed in the text. Other AID cofactors have been observed to regulate AID's functions during DNA deamination by various means via mechanisms that are related and unrelated to that shown above. Further experimentation is required to completely understand AID activity in vivo.
References
-
- Adelman K, Marr MT, Werner J, Saunders A, Ni Z, Andrulis ED, Lis JT 2005. Efficient release from promoter-proximal stall sites requires transcript cleavage factor TFIIS. Mol Cell 17: 103–112 - PubMed
-
- Allen CD, Okada T, Tang HL, Cyster JG 2007b. Imaging of germinal center selection events during affinity maturation. Science 315: 528–531 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources