

Redox control of inflammation in macrophages
- PMID: 23311665
- PMCID: PMC3718318
- DOI: 10.1089/ars.2012.4785
Redox control of inflammation in macrophages
Abstract
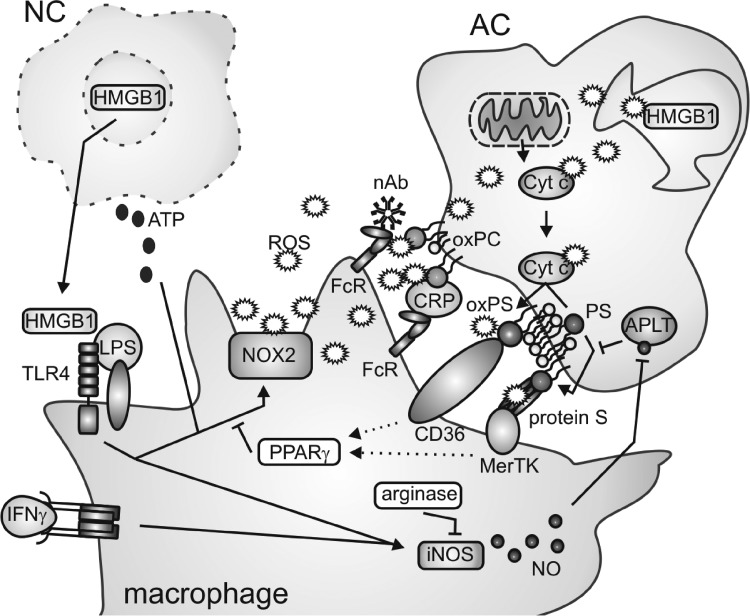
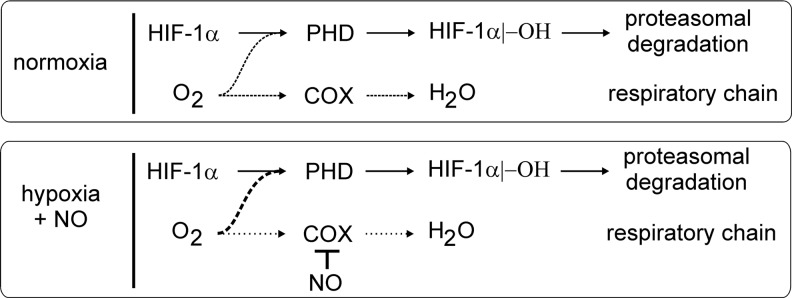
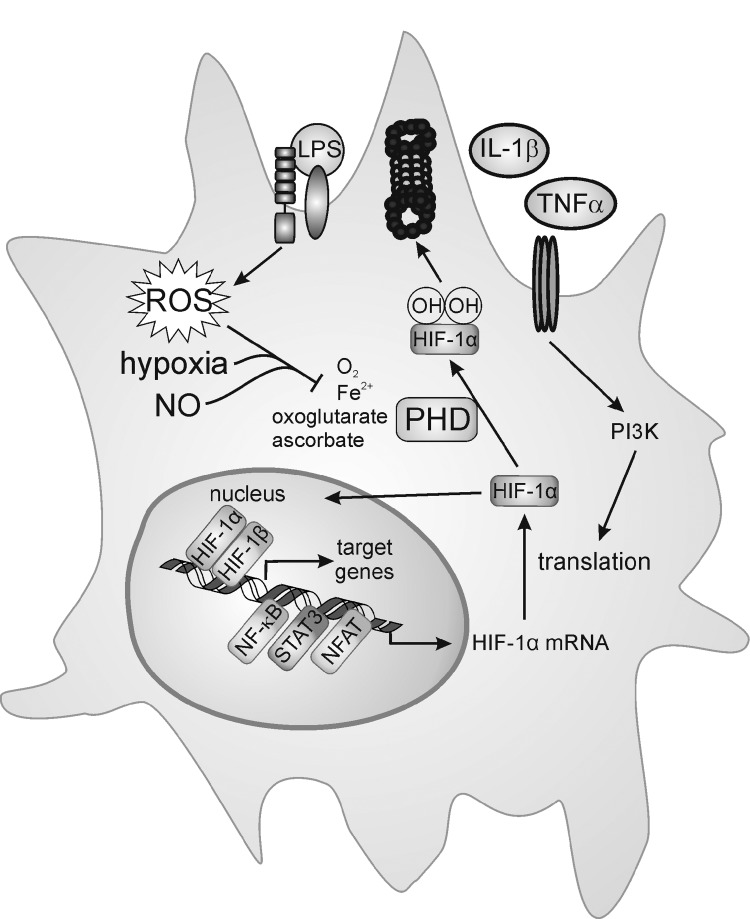
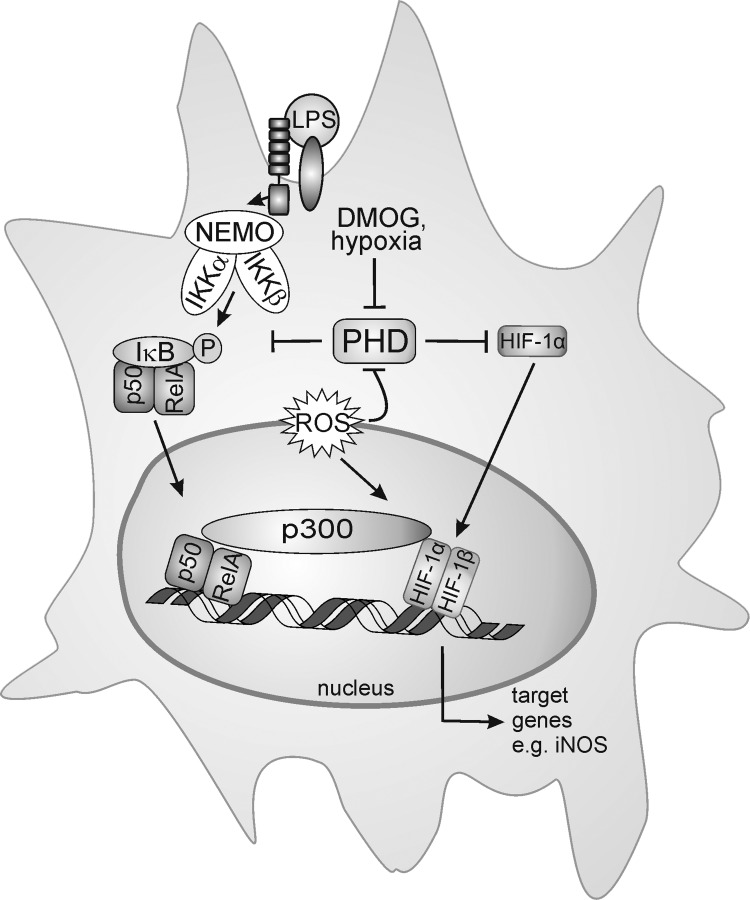
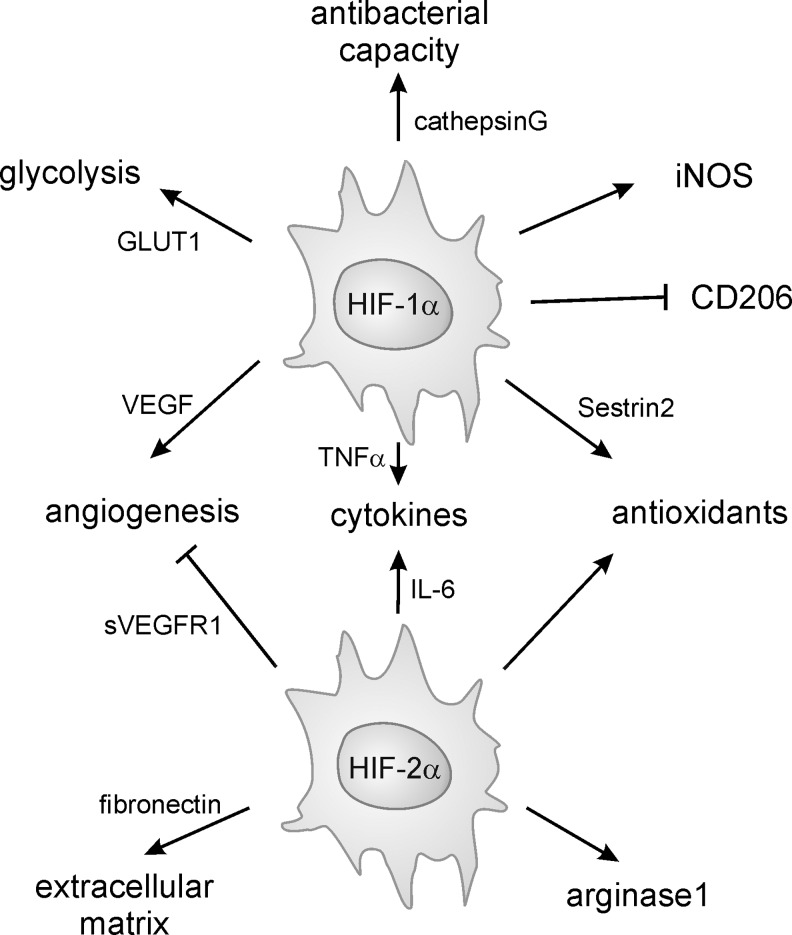
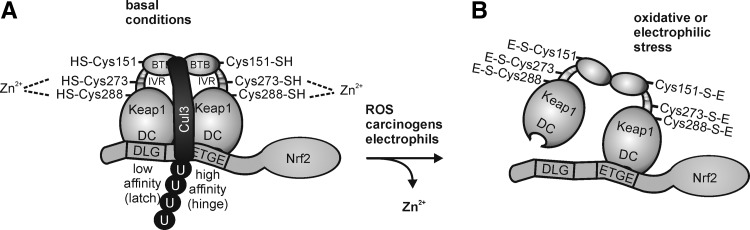
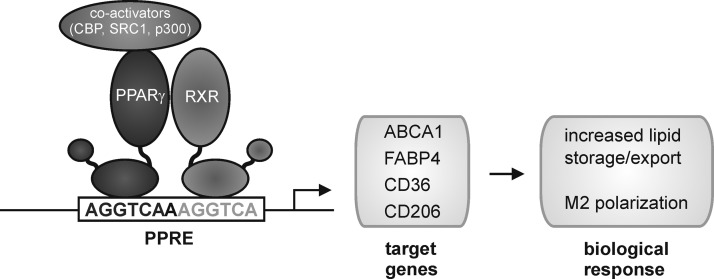
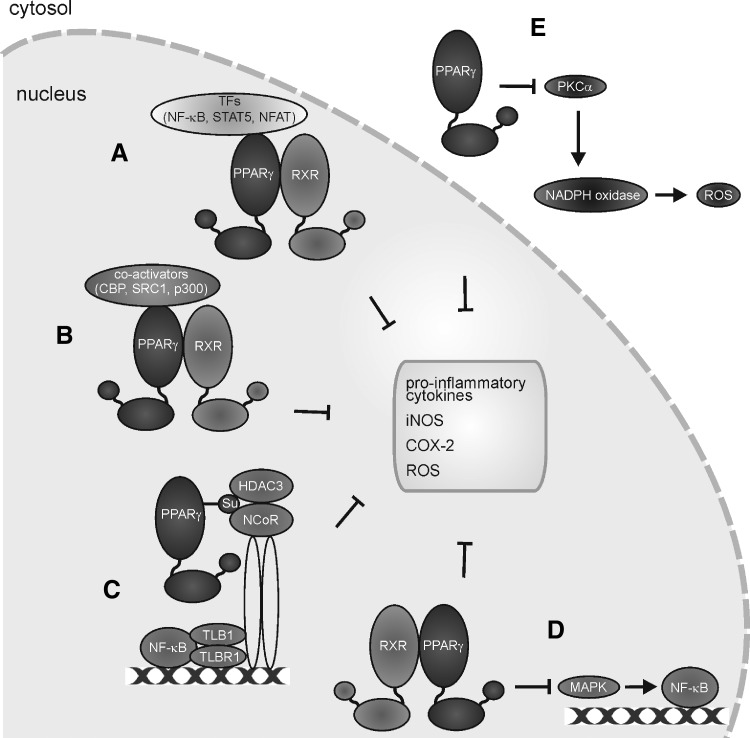
Macrophages are present throughout the human body, constitute important immune effector cells, and have variable roles in a great number of pathological, but also physiological, settings. It is apparent that macrophages need to adjust their activation profile toward a steadily changing environment that requires altering their phenotype, a process known as macrophage polarization. Formation of reactive oxygen species (ROS), derived from NADPH-oxidases, mitochondria, or NO-producing enzymes, are not necessarily toxic, but rather compose a network signaling system, known as redox regulation. Formation of redox signals in classically versus alternatively activated macrophages, their action and interaction at the level of key targets, and the resulting physiology still are insufficiently understood. We review the identity, source, and biological activities of ROS produced during macrophage activation, and discuss how they shape the key transcriptional responses evoked by hypoxia-inducible transcription factors, nuclear-erythroid 2-p45-related factor 2 (Nrf2), and peroxisome proliferator-activated receptor-γ. We summarize the mechanisms how redox signals add to the process of macrophage polarization and reprogramming, how this is controlled by the interaction of macrophages with their environment, and addresses the outcome of the polarization process in health and disease. Future studies need to tackle the option whether we can use the knowledge of redox biology in macrophages to shape their mediator profile in pathophysiology, to accelerate healing in injured tissue, to fight the invading pathogens, or to eliminate settings of altered self in tumors.
Figures
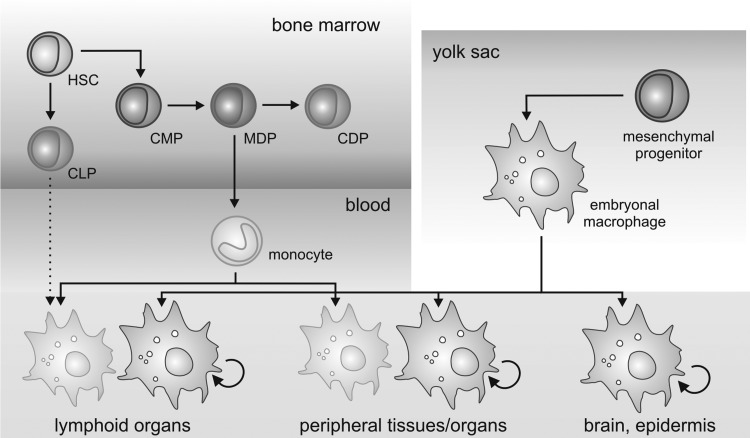
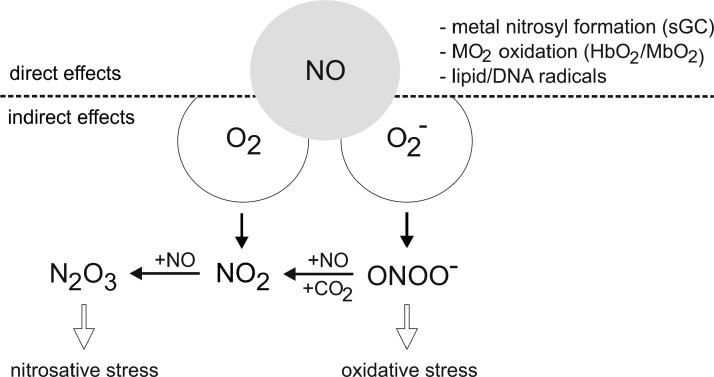
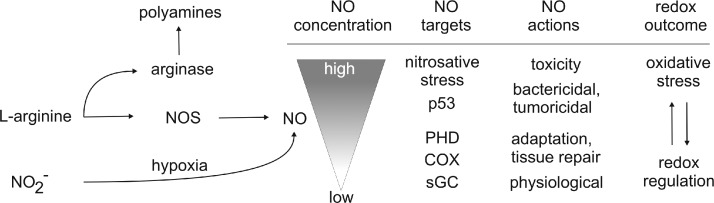
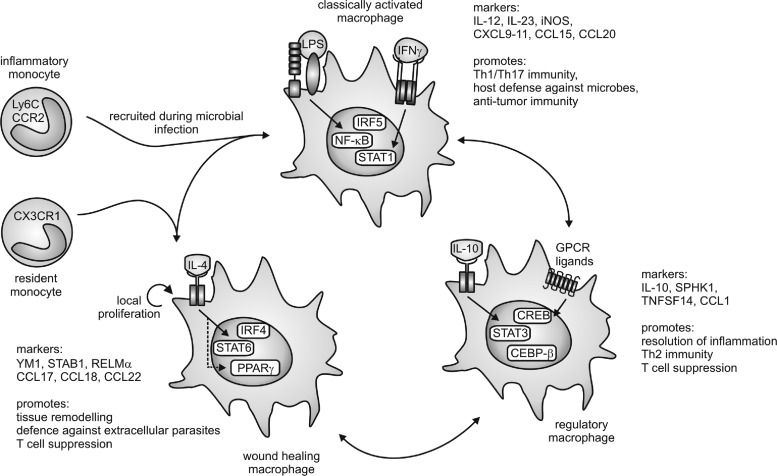



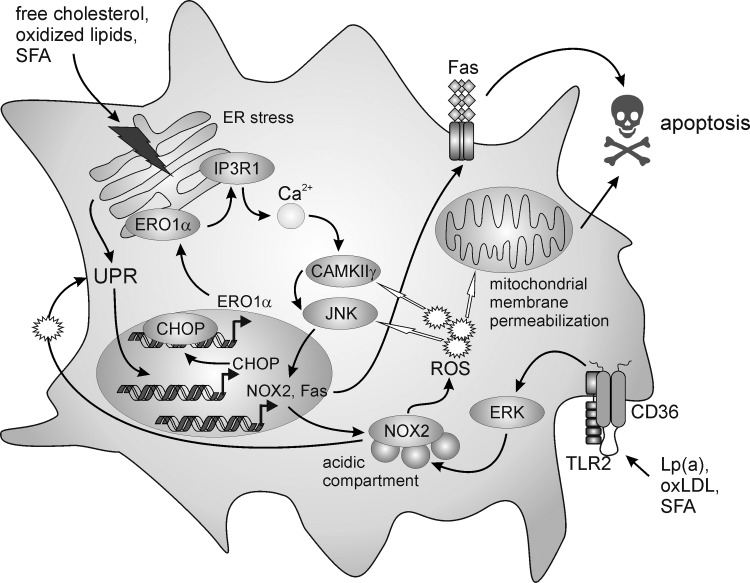
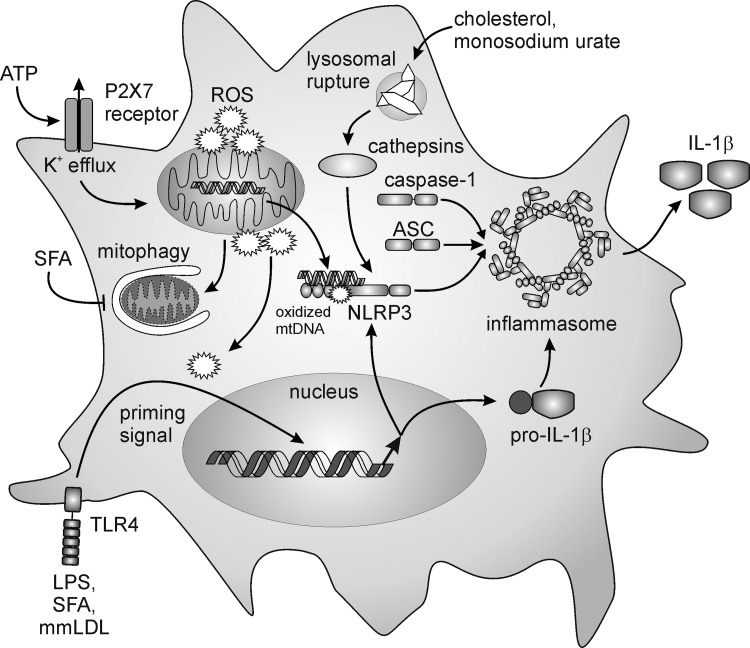
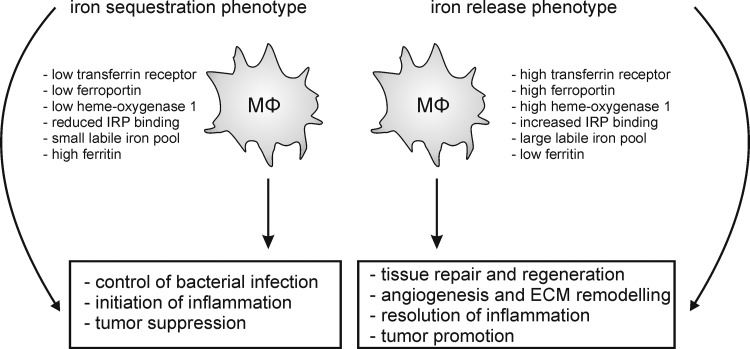
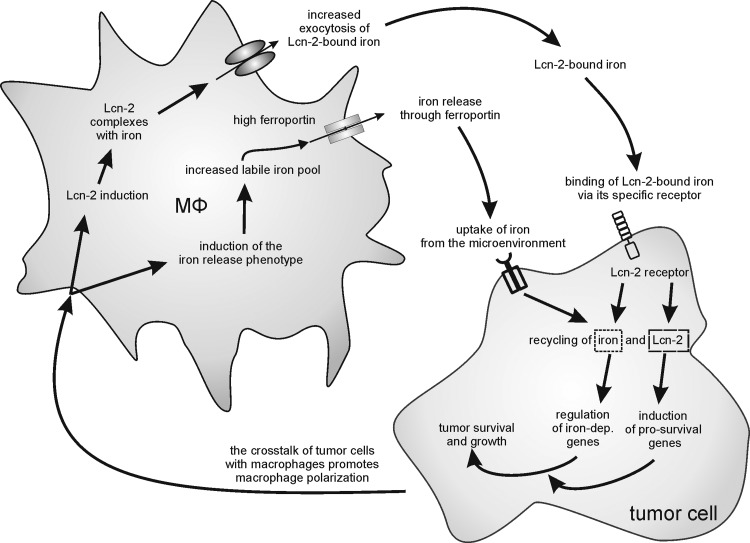
References
-
- Apetoh L. Ghiringhelli F. Tesniere A. Criollo A. Ortiz C. Lidereau R. Mariette C. Chaput N. Mira JP. Delaloge S. Andre F. Tursz T. Kroemer G. Zitvogel L. The interaction between HMGB1 and TLR4 dictates the outcome of anticancer chemotherapy and radiotherapy. Immunol Rev. 2007;220:47–59. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
