

Modeling cognitive dysfunction in neurofibromatosis-1
- PMID: 23312374
- PMCID: PMC3622809
- DOI: 10.1016/j.tins.2012.12.002
Modeling cognitive dysfunction in neurofibromatosis-1
Abstract
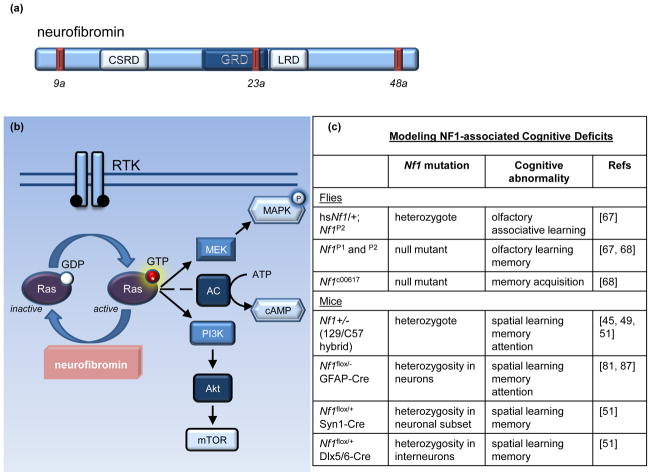
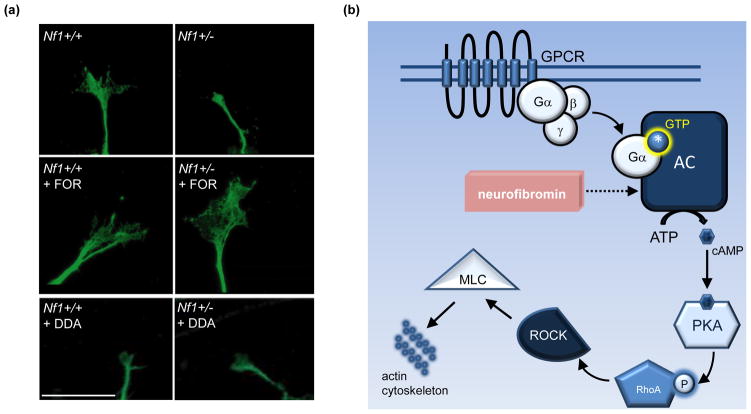
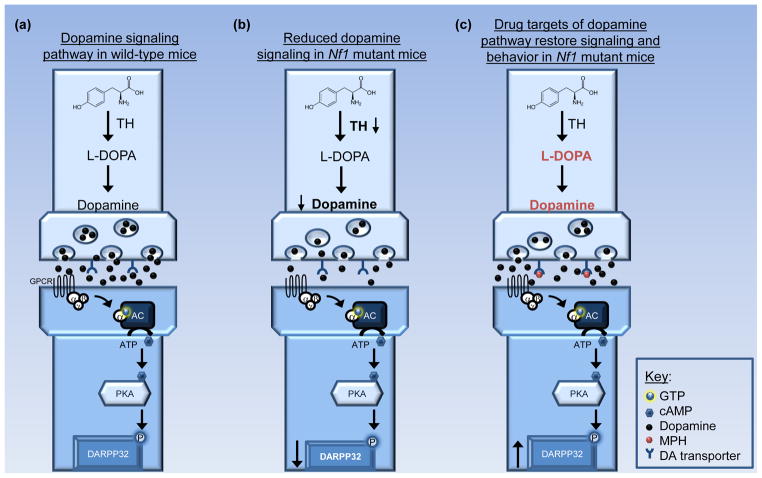
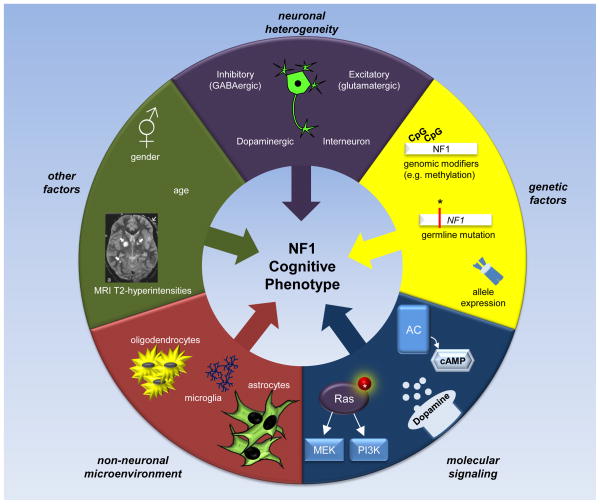
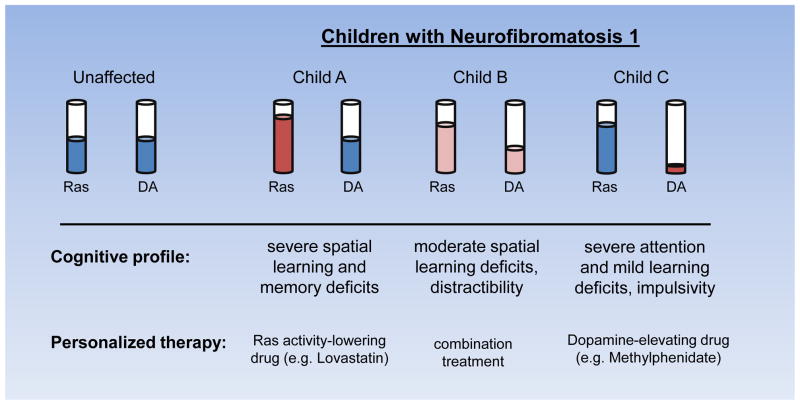
Cognitive dysfunction, including significant impairments in learning, behavior, and attention, is found in over 10% of children in the general population. However, in the common inherited cancer predisposition syndrome, neurofibromatosis type 1 (NF1), the prevalence of these cognitive deficits approaches 70%. As a monogenic disorder, NF1 provides a unique genetic tool to identify and dissect mechanistically the molecular and cellular bases underlying cognitive dysfunction. In this review, we discuss Nf1 fly and mouse systems that mimic many of the cognitive abnormalities seen in children with NF1. Further, we describe discoveries from these models that have uncovered defects in the regulation of Ras activity, cAMP generation, and dopamine homeostasis as key mechanisms important for cognitive dysfunction in children with NF1.
Copyright © 2012 Elsevier Ltd. All rights reserved.
Figures
References
-
- Hyman SL, et al. The nature and frequency of cognitive deficits in children with neurofibromatosis type 1. Neurology. 2005;65:1037–1044. - PubMed
-
- Mautner VF, et al. Treatment of ADHD in neurofibromatosis type 1. Developmental medicine and child neurology. 2002;44:164–170. - PubMed
-
- Hyman SL, et al. Learning disabilities in children with neurofibromatosis type 1: subtypes, cognitive profile, and attention-deficit-hyperactivity disorder. Developmental medicine and child neurology. 2006;48:973–977. - PubMed
-
- Ferner RE. The neurofibromatoses. Practical neurology. 2010;10:82–93. - PubMed
-
- Wallace MR, et al. Type 1 neurofibromatosis gene: identification of a large transcript disrupted in three NF1 patients. Science. 1990;249:181–186. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
