

Wnt your brain be inflamed? Yes, it Wnt!
- PMID: 23312954
- PMCID: PMC3595301
- DOI: 10.1016/j.molmed.2012.12.001
Wnt your brain be inflamed? Yes, it Wnt!
Abstract
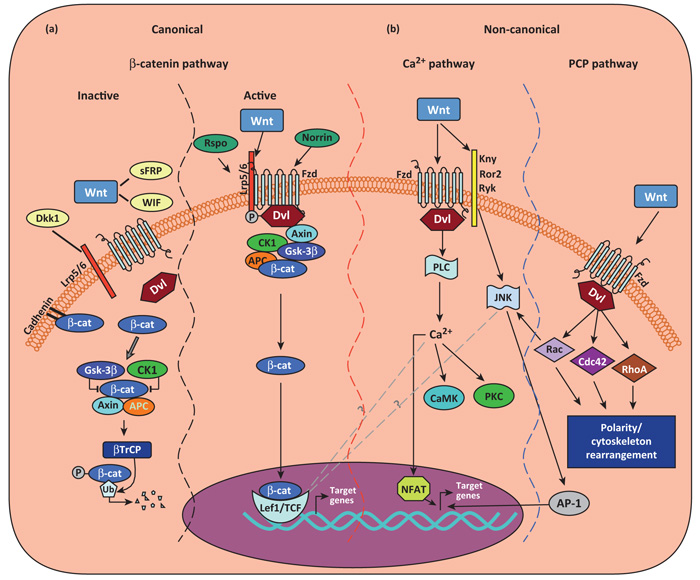
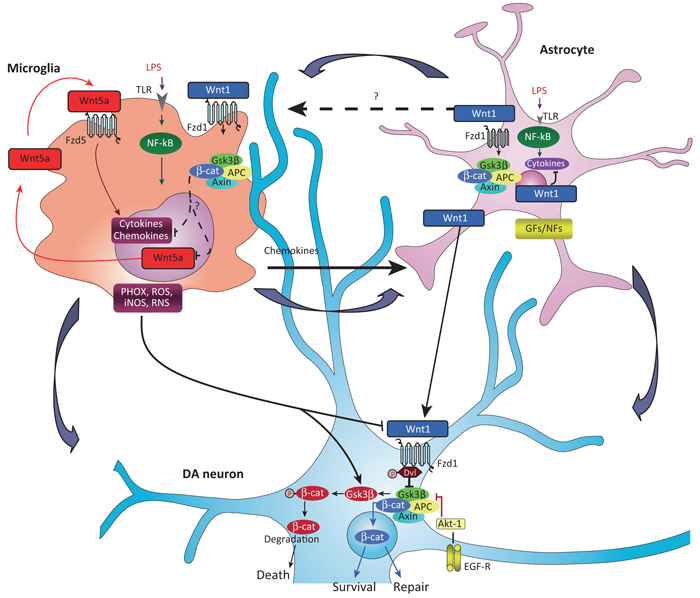
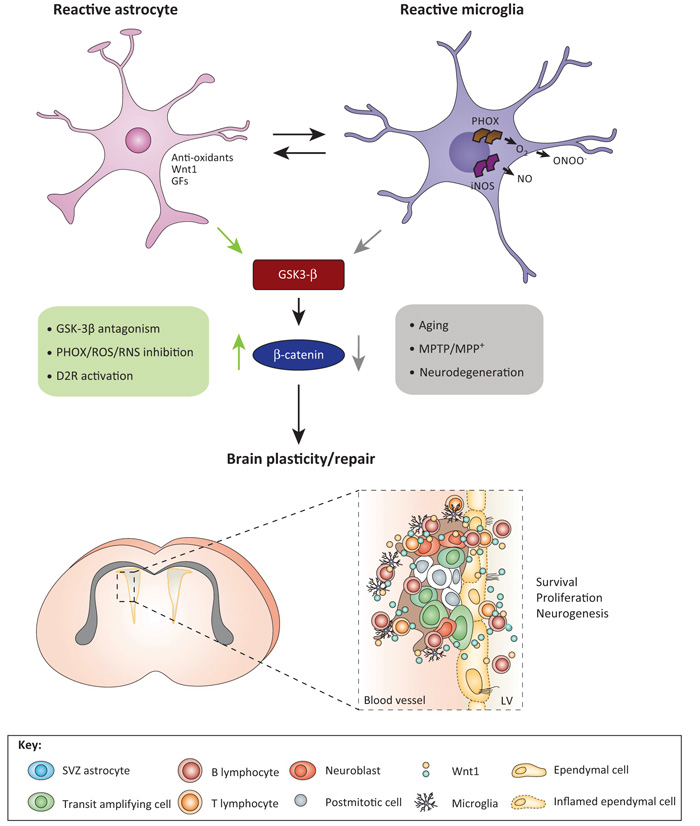
The roles of Wnts in neural development, synaptogenesis, and cancer are generally well characterized. Nonetheless, evidence exists that interactions between the immune and nervous systems control major brain regenerative processes ranging from physiological or pathological (reparative) regeneration to neurogenesis and synaptic plasticity. Recent studies describe deregulated Wnt-Fzd signaling in degenerative and inflammatory central nervous system (CNS) disorders, and the expression of Wnt signaling components in the immune system, and in immune-like cells of the mammalian CNS. This would suggest a likely involvement of Wnts in inflammation-driven brain damage and inflammation-directed brain repair. Here, we review how Wnts modulate neuroimmune interactions and offer a perspective on the most challenging therapeutic opportunities for those CNS diseases where injury-reactive Wnt-flavored inflammation precedes secondary neurodegeneration.
Copyright © 2012 Elsevier Ltd. All rights reserved.
Figures
Similar articles
-
Wnt signaling in development and disease.Neurobiol Dis. 2010 May;38(2):148-53. doi: 10.1016/j.nbd.2009.09.003. Epub 2009 Sep 16. Neurobiol Dis. 2010. PMID: 19765659 Free PMC article. Review.
-
Ryks: new partners for Wnts in the developing and regenerating nervous system.Trends Neurosci. 2010 Feb;33(2):84-92. doi: 10.1016/j.tins.2009.11.005. Epub 2009 Dec 11. Trends Neurosci. 2010. PMID: 20004982 Review.
-
Emerging roles of Wnts in the adult nervous system.Nat Rev Neurosci. 2010 Feb;11(2):77-86. doi: 10.1038/nrn2755. Epub 2009 Dec 16. Nat Rev Neurosci. 2010. PMID: 20010950 Review.
-
Wnt signaling during synaptic development and plasticity.Curr Opin Neurobiol. 2011 Feb;21(1):151-9. doi: 10.1016/j.conb.2010.12.002. Epub 2011 Jan 14. Curr Opin Neurobiol. 2011. PMID: 21239163 Free PMC article. Review.
-
Glial development: the crossroads of regeneration and repair in the CNS.Neuron. 2014 Jul 16;83(2):283-308. doi: 10.1016/j.neuron.2014.06.010. Neuron. 2014. PMID: 25033178 Free PMC article. Review.
Cited by
-
Lithium and Atypical Antipsychotics: The Possible WNT/β Pathway Target in Glaucoma.Biomedicines. 2021 Apr 26;9(5):473. doi: 10.3390/biomedicines9050473. Biomedicines. 2021. PMID: 33925885 Free PMC article. Review.
-
Interplay of Opposing Effects of the WNT/β-Catenin Pathway and PPARγ and Implications for SARS-CoV2 Treatment.Front Immunol. 2021 Apr 13;12:666693. doi: 10.3389/fimmu.2021.666693. eCollection 2021. Front Immunol. 2021. PMID: 33927728 Free PMC article. Review.
-
FoxO Transcription Factors and Regenerative Pathways in Diabetes Mellitus.Curr Neurovasc Res. 2015;12(4):404-13. doi: 10.2174/1567202612666150807112524. Curr Neurovasc Res. 2015. PMID: 26256004 Free PMC article. Review.
-
Molecular mechanisms of developmental pathways in neurological disorders: a pharmacological and therapeutic review.Open Biol. 2022 Mar;12(3):210289. doi: 10.1098/rsob.210289. Epub 2022 Mar 16. Open Biol. 2022. PMID: 35291879 Free PMC article. Review.
-
Thermodynamics in Neurodegenerative Diseases: Interplay Between Canonical WNT/Beta-Catenin Pathway-PPAR Gamma, Energy Metabolism and Circadian Rhythms.Neuromolecular Med. 2018 Jun;20(2):174-204. doi: 10.1007/s12017-018-8486-x. Epub 2018 Mar 23. Neuromolecular Med. 2018. PMID: 29572723 Review.
References
-
- Toledo EM, et al. Wnt signaling in neuroprotection and stem cell differentiation. Prog. Neurobiol. 2008;86:281–296. - PubMed
-
- Mastroiacovo F, et al. Induction of the Wnt antagonist, Dickkopf-1, contributes to the development of neuronal death in models of brain focal ischemia. J. Cereb. Blood Flow Metab. 2009;29:264–276. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
