

Direct conversion of fibroblasts to neurons by reprogramming PTB-regulated microRNA circuits
- PMID: 23313552
- PMCID: PMC3552026
- DOI: 10.1016/j.cell.2012.11.045
Direct conversion of fibroblasts to neurons by reprogramming PTB-regulated microRNA circuits
Abstract
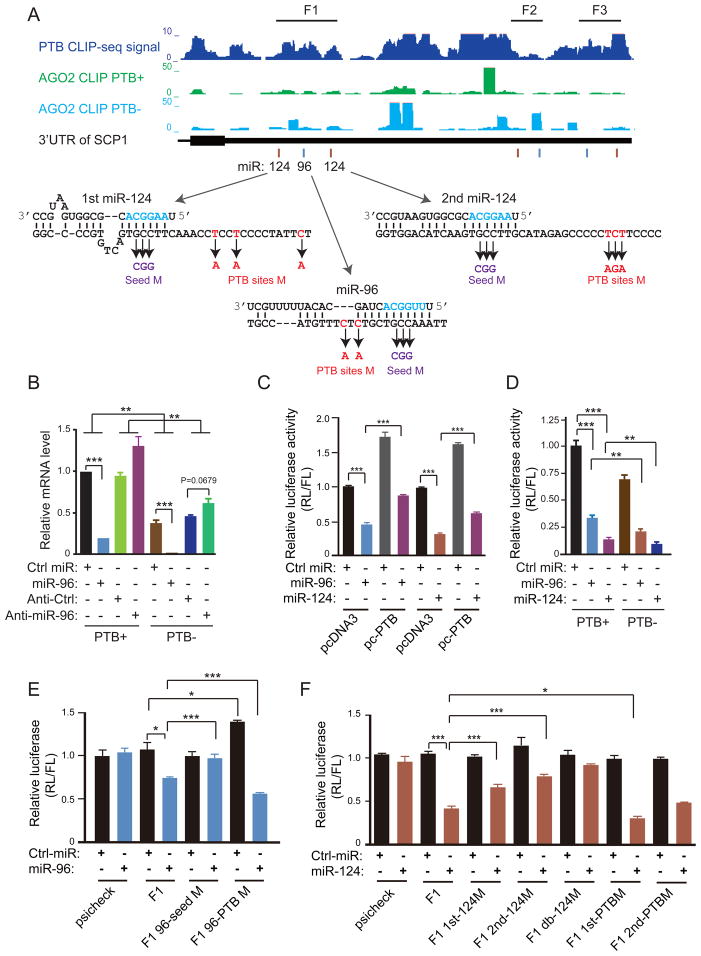
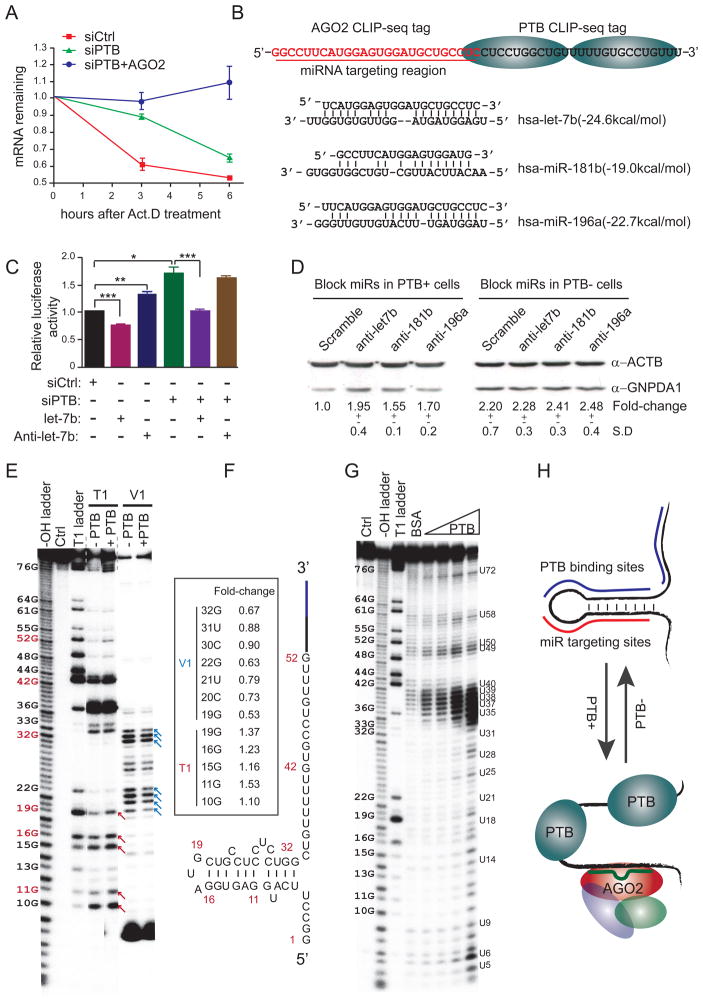
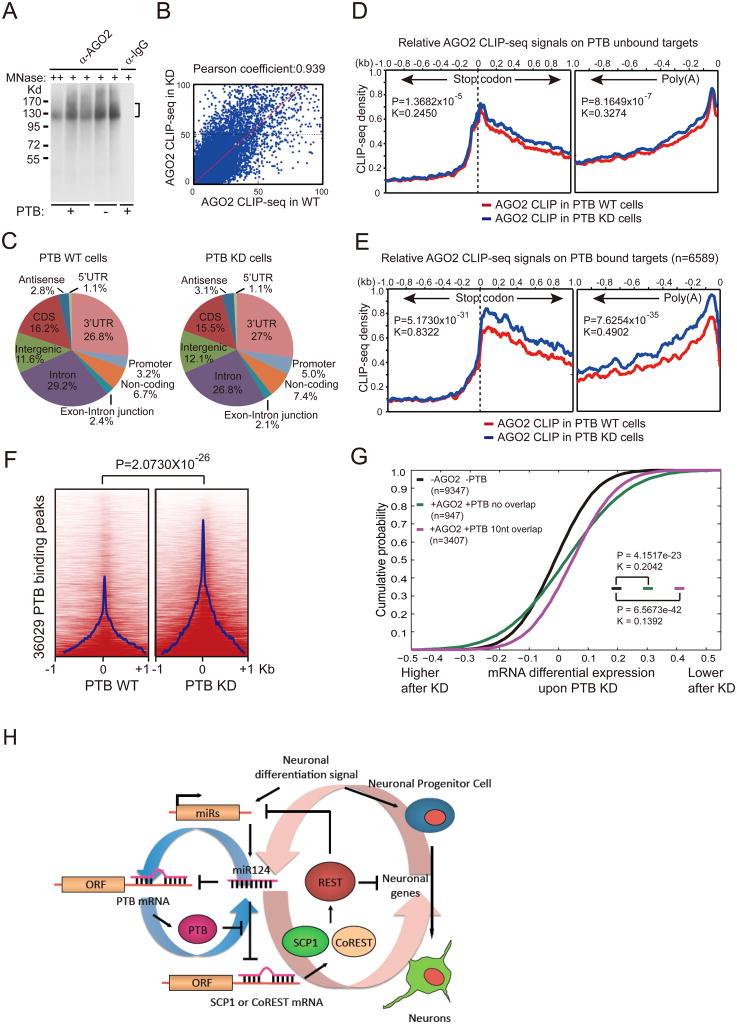
The induction of pluripotency or trans-differentiation of one cell type to another can be accomplished with cell-lineage-specific transcription factors. Here, we report that repression of a single RNA binding polypyrimidine-tract-binding (PTB) protein, which occurs during normal brain development via the action of miR-124, is sufficient to induce trans-differentiation of fibroblasts into functional neurons. Besides its traditional role in regulated splicing, we show that PTB has a previously undocumented function in the regulation of microRNA functions, suppressing or enhancing microRNA targeting by competitive binding on target mRNA or altering local RNA secondary structure. A key event during neuronal induction is the relief of PTB-mediated blockage of microRNA action on multiple components of the REST complex, thereby derepressing a large array of neuronal genes, including miR-124 and multiple neuronal-specific transcription factors, in nonneuronal cells. This converts a negative feedback loop to a positive one to elicit cellular reprogramming to the neuronal lineage.
Copyright © 2013 Elsevier Inc. All rights reserved.
Figures
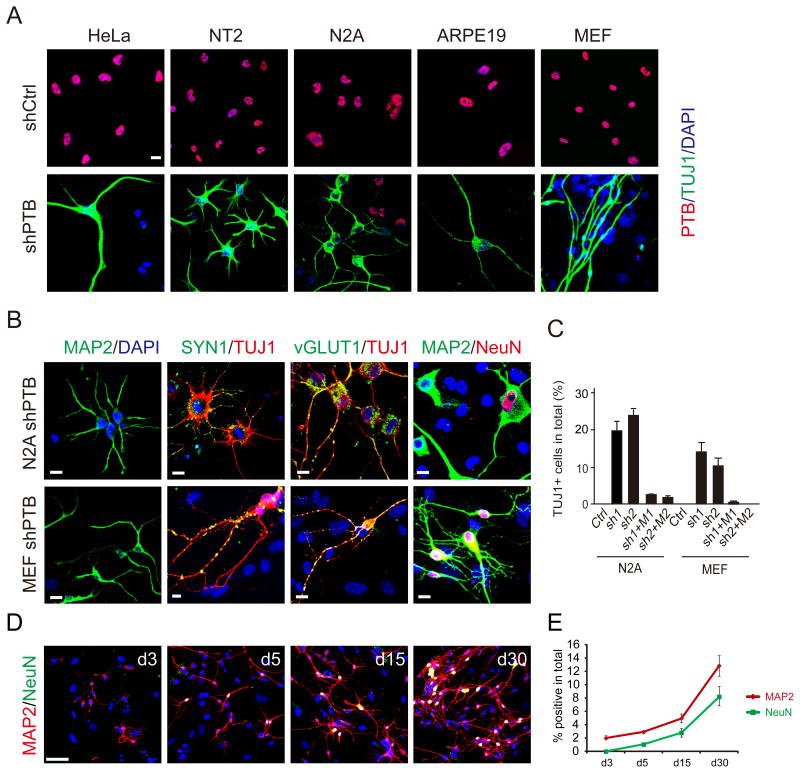
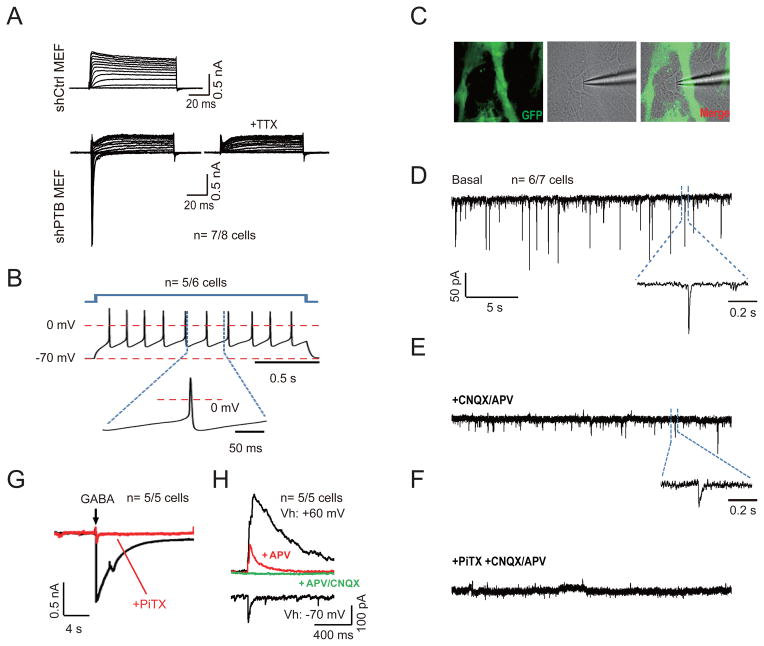
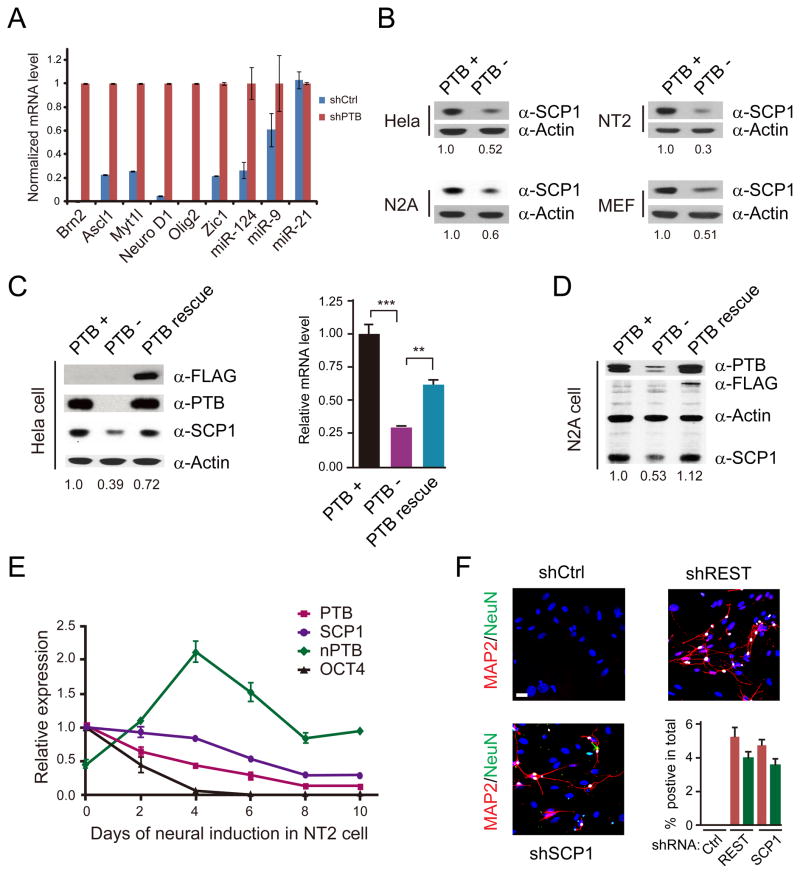
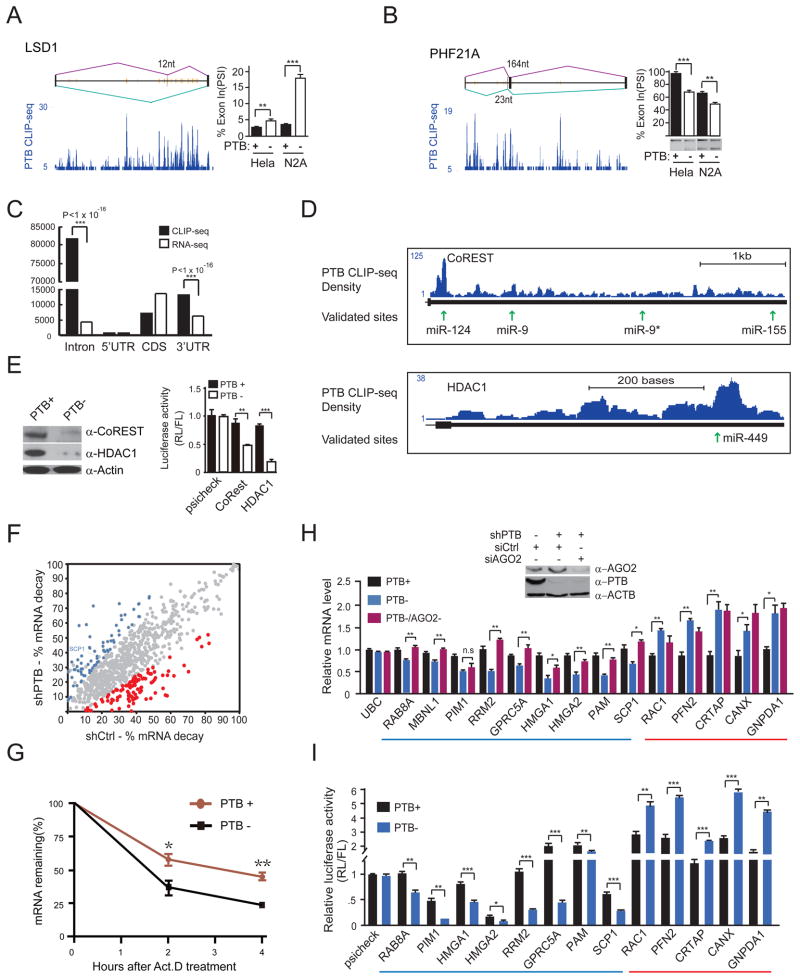
References
-
- Aoki H, Hara A, Era T, Kunisada T, Yamada Y. Genetic ablation of Rest leads to in vitro-specific derepression of neuronal genes during neurogenesis. Development. 2012;139:667–677. - PubMed
-
- Auld KL, Silver PA. Transcriptional regulation by the proteasome as a mechanism for cellular protein homeostasis. Cell Cycle. 2006;5:1503–1505. - PubMed
-
- Ballas N, Grunseich C, Lu DD, Speh JC, Mandel G. REST and its corepressors mediate plasticity of neuronal gene chromatin throughout neurogenesis. Cell. 2005;121:645–657. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
