

Bioinformatics analysis of large-scale viral sequences: from construction of data sets to annotation of a phylogenetic tree
- PMID: 23314574
- PMCID: PMC3544756
- DOI: 10.4161/viru.23161
Bioinformatics analysis of large-scale viral sequences: from construction of data sets to annotation of a phylogenetic tree
Abstract
Due to a significant decrease in the cost of DNA sequencing, the number of sequences submitted to the public databases has dramatically increased in recent years. Efficient analysis of these data sets may lead to a significant understanding of the nature of pathogens such as bacteria, viruses, parasites, etc. However, this has raised questions about the efficacy of currently available algorithms for the study of pathogen evolution and construction of phylogenetic trees. While the advanced algorithms and corresponding programs are being developed, it is crucial to optimize the available ones in order to cope with the current need. The protocol presented in this study is optimized using a number of strategies currently being proposed for handling large-scale DNA sequence data sets, and offers a highly efficacious and accurate method for computing phylogenetic trees with limited computer resources. The protocol may take up to 36 h for construction and annotation of a final tree of about 20,000 sequences.
Figures
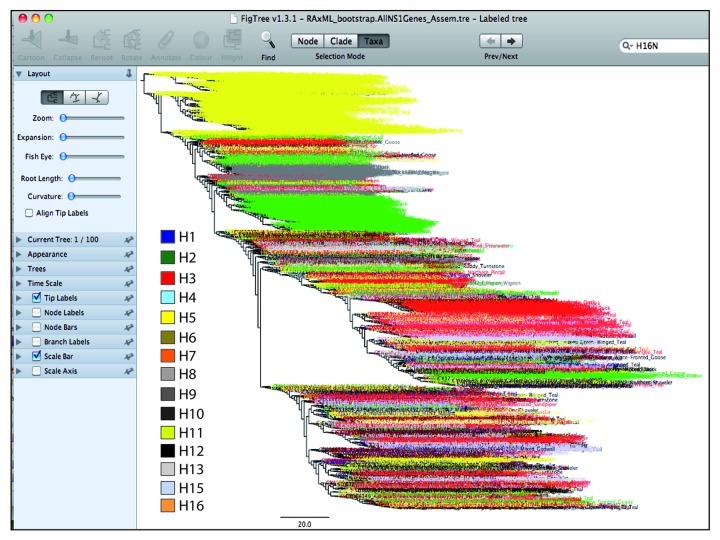
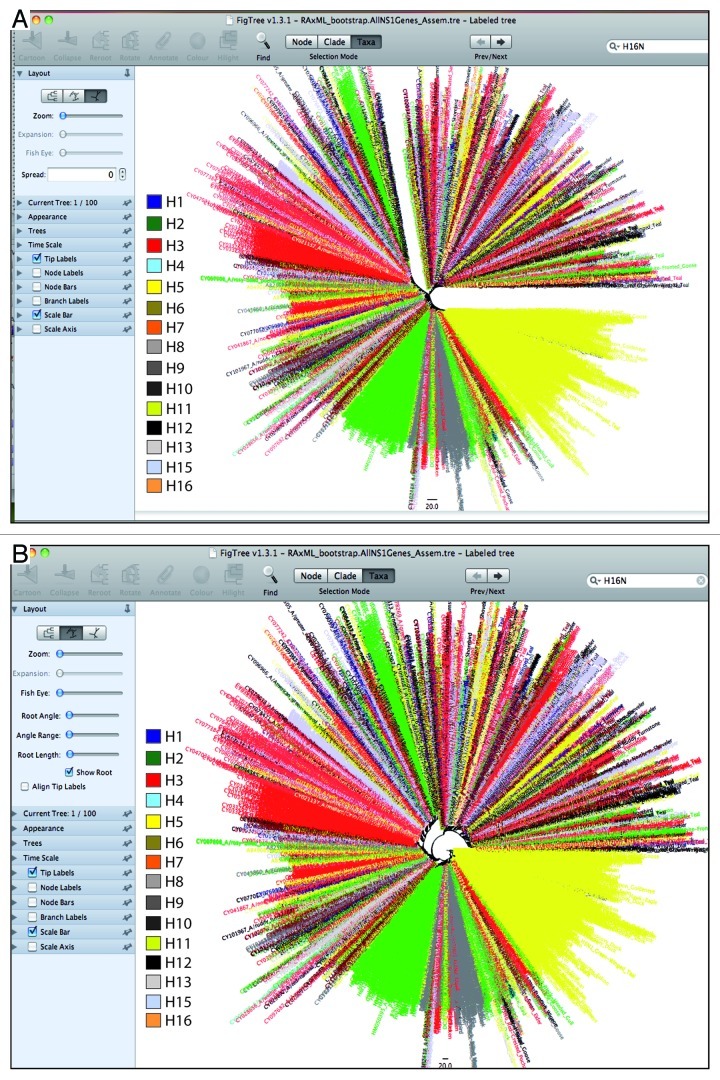
References
-
- Swofford D, Olsen G, Waddel P, Hillis DM. Phylogenetic inference. Pages in (Molecular systematics, 2nd edition (D. M. Hillis, C.Moritz, and B. K. Mable, eds.). Sinauer, Sunderland, Massachusetts.
-
- Page R, Holmes E. Molecular evolution: A phylogenetic approach. Blackwell, Osney Mead, Oxford, UK.
-
- Jin L, Nei M. Limitations of the evolutionary parsimony method of phylogenetic analysis. Mol Biol Evol. 1990;7:82–102. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources