

Structure-activity relationship study reveals ML240 and ML241 as potent and selective inhibitors of p97 ATPase
- PMID: 23316025
- PMCID: PMC3662613
- DOI: 10.1002/cmdc.201200520
Structure-activity relationship study reveals ML240 and ML241 as potent and selective inhibitors of p97 ATPase
Abstract
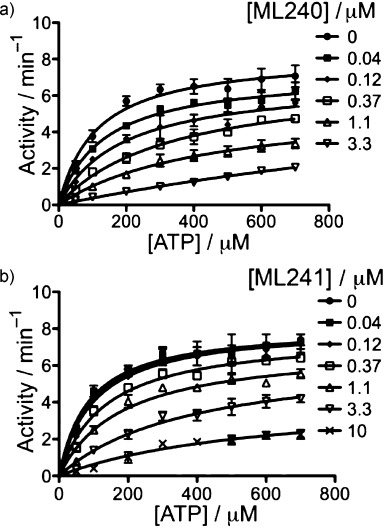
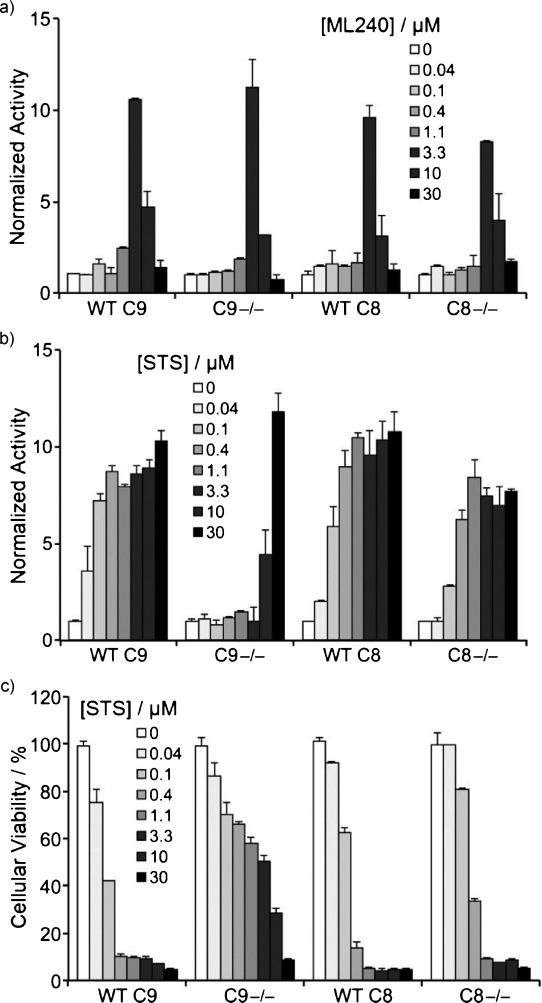
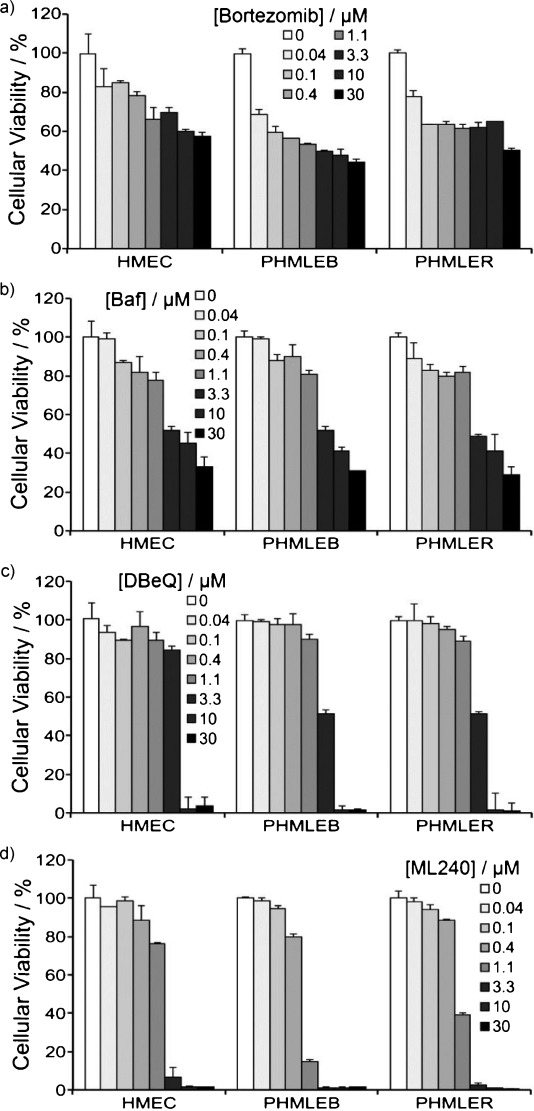
To discover more potent p97 inhibitors, we carried out a structure-activity relationship study of the quinazoline scaffold previously identified from our HTS campaigns. Two improved inhibitors, ML240 and ML241, inhibit p97 ATPase with IC(50) values of 100 nM. Both compounds inhibited degradation of a p97-dependent but not a p97-independent proteasome substrate in a dual-reporter cell line. They also impaired the endoplasmic-reticulum-associated degradation (ERAD) pathway. Unexpectedly, ML240 potently stimulated accumulation of LC3-II within minutes, inhibited cancer cell growth, and rapidly mobilized the executioner caspases 3 and 7, whereas ML241 did not. The behavior of ML240 suggests that disruption of the protein homeostasis function of p97 leads to more rapid activation of apoptosis than is observed with a proteasome inhibitor. Further characterization revealed that ML240 has broad antiproliferative activity toward the NCI-60 panel of cancer cell lines, but slightly lower activity toward normal cells. ML240 also synergizes with the proteasome inhibitor MG132 to kill multiple colon cancer cell lines. Meanwhile, both probes have low off-target activity toward a panel of protein kinases and central nervous system targets. Our results nominate ML240 as a promising starting point for the development of a novel agent for the chemotherapy of cancer, and provide a rationale for developing pathway-specific p97 inhibitors.
Copyright © 2013 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.
Figures







References
-
- Giaever G, et al. Nature. 2002;418:387–391. - PubMed
-
- Müller JM, Deinhardt K, Rosewell I, Warren G, Shima DT. Biochem. Biophys. Res. Commun. 2007;354:459–465. - PubMed
-
- Kondo H, Rabouille C, Newman R, Levine TP, Pappin D, Freemont P, Warren G. Nature. 1997;388:75–78. - PubMed
-
- Golbik R, Lupas AN, Koretke KK, Baumeister W, Peters J. Biol. Chem. 1999;380:1049–1062. - PubMed
-
- Rabouille C, Levine TP, Peters JM, Warren G. Cell. 1995;82:905–914. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
