

Hydrogen sulfide stimulates ischemic vascular remodeling through nitric oxide synthase and nitrite reduction activity regulating hypoxia-inducible factor-1α and vascular endothelial growth factor-dependent angiogenesis
- PMID: 23316304
- PMCID: PMC3541625
- DOI: 10.1161/JAHA.112.004093
Hydrogen sulfide stimulates ischemic vascular remodeling through nitric oxide synthase and nitrite reduction activity regulating hypoxia-inducible factor-1α and vascular endothelial growth factor-dependent angiogenesis
Abstract
Background: Hydrogen sulfide (H(2)S) therapy is recognized as a modulator of vascular function during tissue ischemia with the notion of potential interactions of nitric oxide (NO) metabolism. However, little is known about specific biochemical mechanisms or the importance of H(2)S activation of NO metabolism during ischemic tissue vascular remodeling. The goal of this study was to determine the effect of H(2)S on NO metabolism during chronic tissue ischemia and subsequent effects on ischemic vascular remodeling responses.
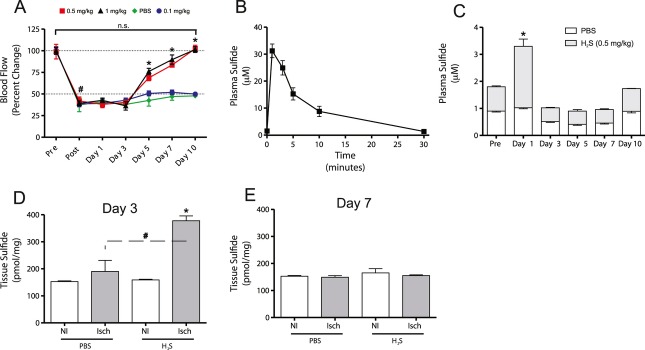
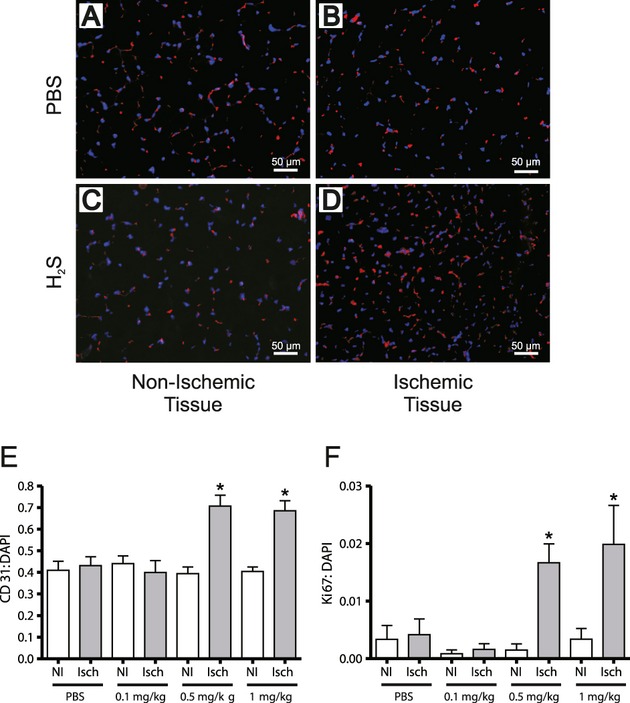
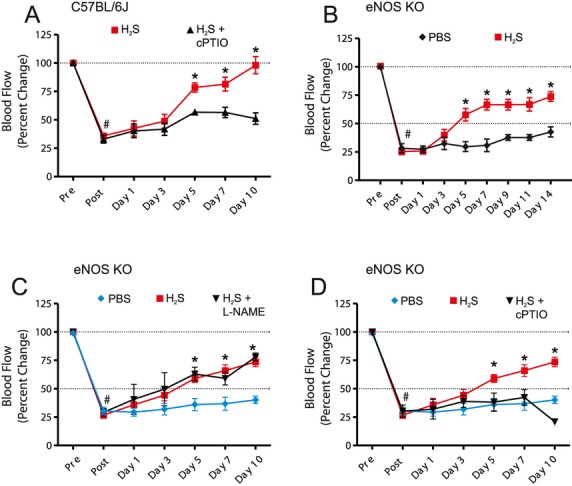
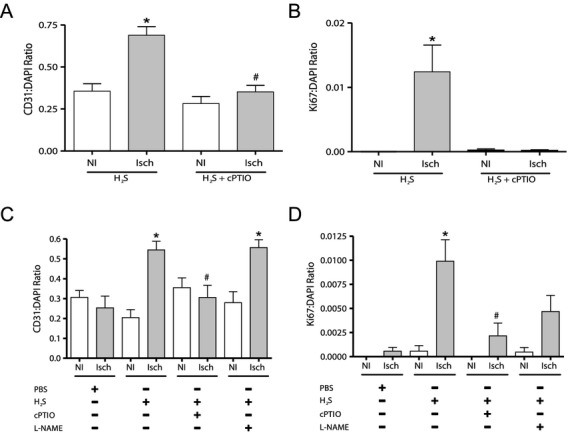
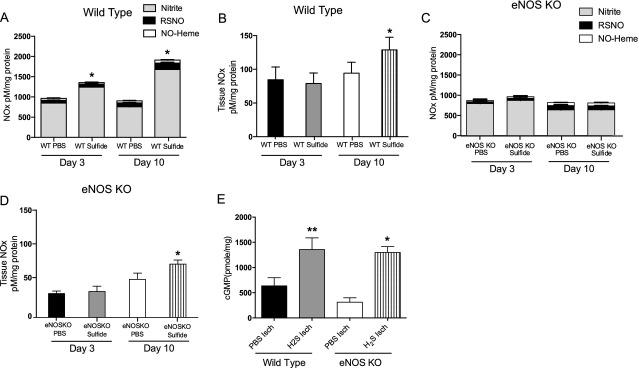
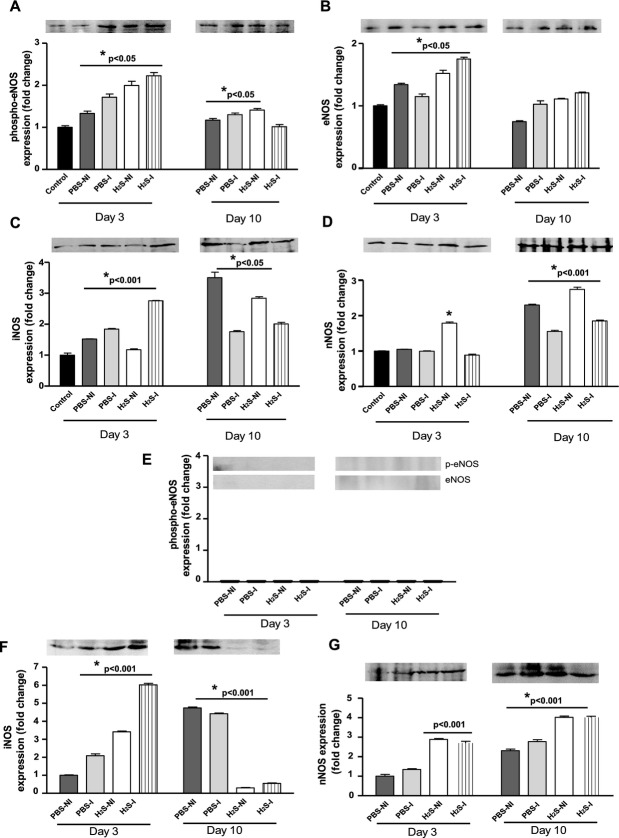
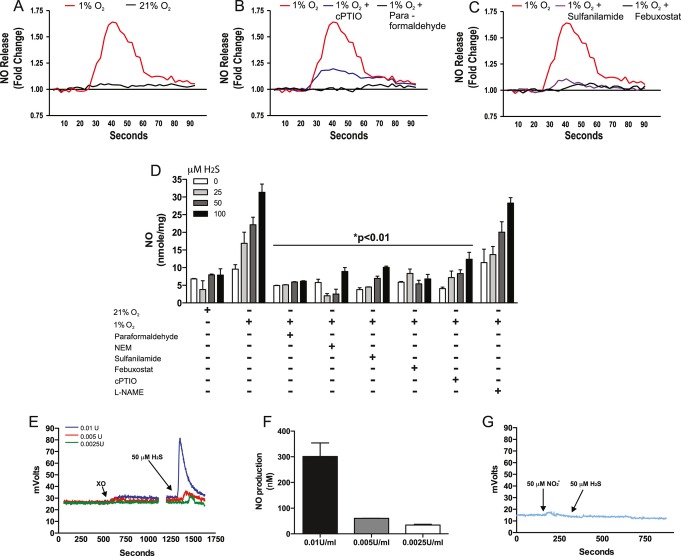
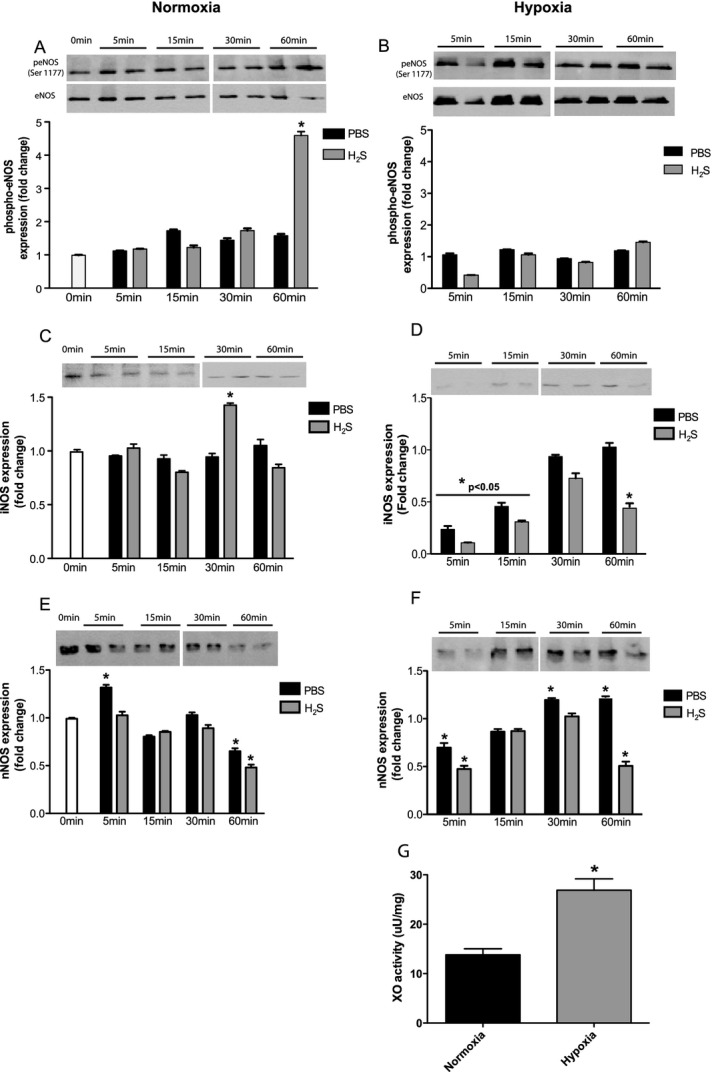
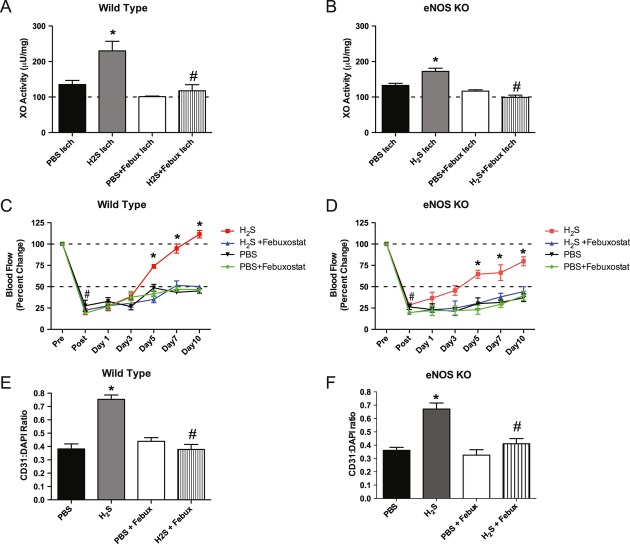
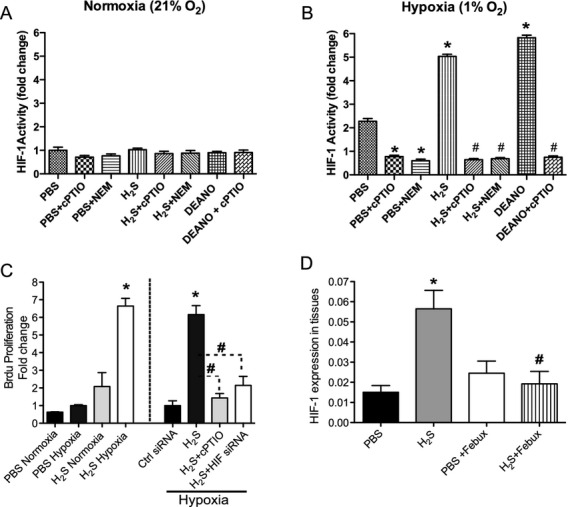
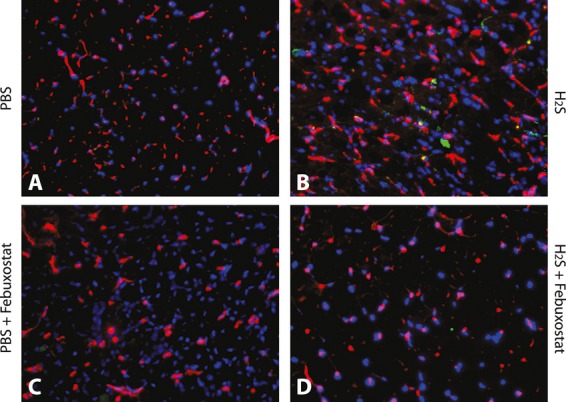
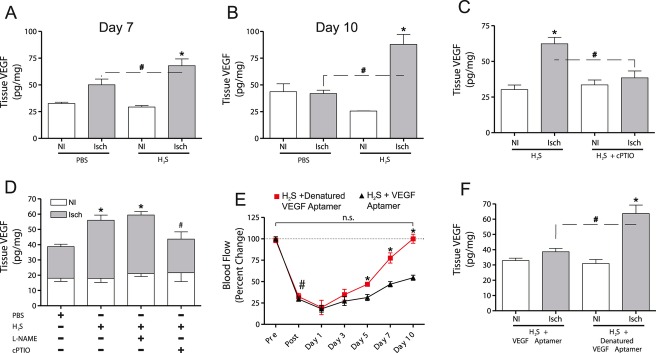
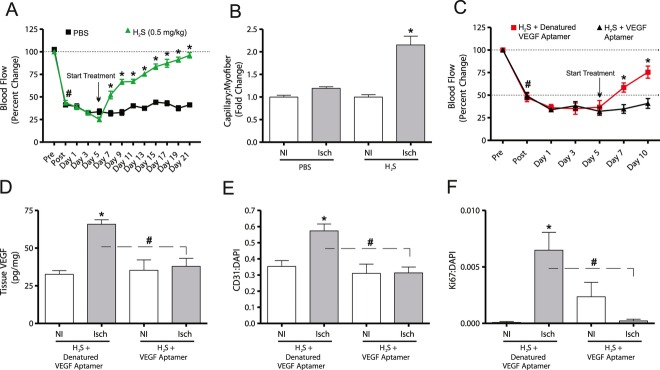
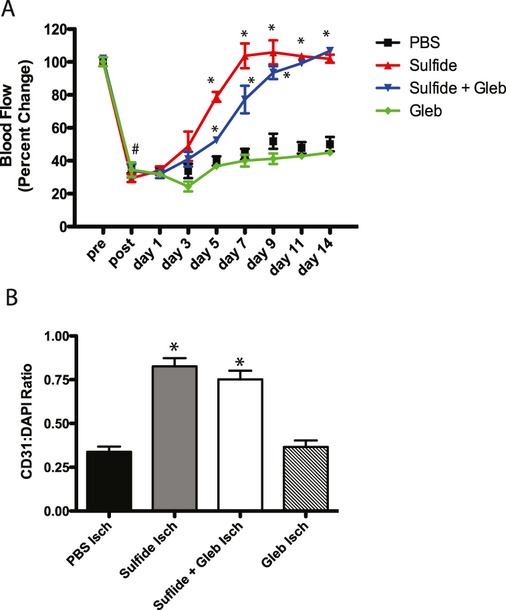
Methods and results: The unilateral, permanent femoral artery ligation model of hind-limb ischemia was performed in C57BL/6J wild-type and endothelial NO synthase-knockout mice to evaluate exogenous H(2)S effects on NO bioavailability and ischemic revascularization. We found that H(2)S selectively restored chronic ischemic tissue function and viability by enhancing NO production involving both endothelial NO synthase and nitrite reduction mechanisms. Importantly, H(2)S increased ischemic tissue xanthine oxidase activity, hind-limb blood flow, and angiogenesis, which were blunted by the xanthine oxidase inhibitor febuxostat. H(2)S treatment increased ischemic tissue and endothelial cell hypoxia-inducible factor-1α expression and activity and vascular endothelial growth factor protein expression and function in a NO-dependent manner that was required for ischemic vascular remodeling.
Conclusions: These data demonstrate that H(2)S differentially regulates NO metabolism during chronic tissue ischemia, highlighting novel biochemical pathways to increase NO bioavailability for ischemic vascular remodeling.
Keywords: angiogenesis; ischemia; nitric oxide; vascular endothelial growth factor; xanthine oxidase.
Figures
References
-
- Petersen LC. The effect of inhibitors on the oxygen kinetics of cytochrome c oxidase. Biochim Biophys Acta.. 1977;460:299-307. - PubMed
-
- Smith RP, Gosselin RE. Hydrogen sulfide poisoning. J Occup Med.. 1979;21:93-97. - PubMed
-
- Guidotti TL. Hydrogen sulphide. Occup Med (Lond).. 1996;46:367-371. - PubMed
-
- Tang C, Li X, Du J. Hydrogen sulfide as a new endogenous gaseous transmitter in the cardiovascular system. Curr Vasc Pharmacol.. 2006;4:17-22. - PubMed
-
- Kamoun P. Endogenous production of hydrogen sulfide in mammals. Amino Acids.. 2004;26:243-254. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
