

Random generalized linear model: a highly accurate and interpretable ensemble predictor
- PMID: 23323760
- PMCID: PMC3645958
- DOI: 10.1186/1471-2105-14-5
Random generalized linear model: a highly accurate and interpretable ensemble predictor
Abstract
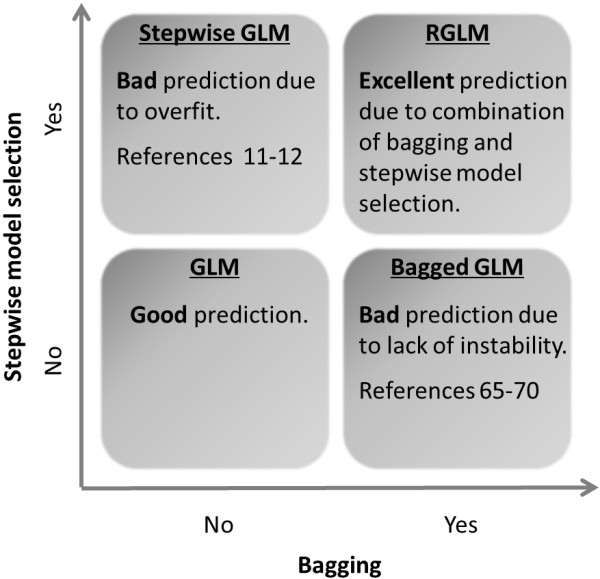
Background: Ensemble predictors such as the random forest are known to have superior accuracy but their black-box predictions are difficult to interpret. In contrast, a generalized linear model (GLM) is very interpretable especially when forward feature selection is used to construct the model. However, forward feature selection tends to overfit the data and leads to low predictive accuracy. Therefore, it remains an important research goal to combine the advantages of ensemble predictors (high accuracy) with the advantages of forward regression modeling (interpretability). To address this goal several articles have explored GLM based ensemble predictors. Since limited evaluations suggested that these ensemble predictors were less accurate than alternative predictors, they have found little attention in the literature.
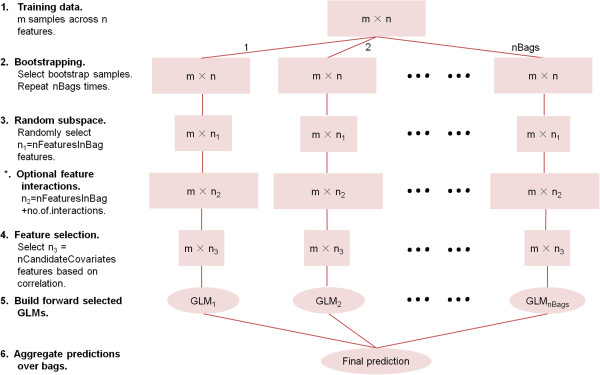
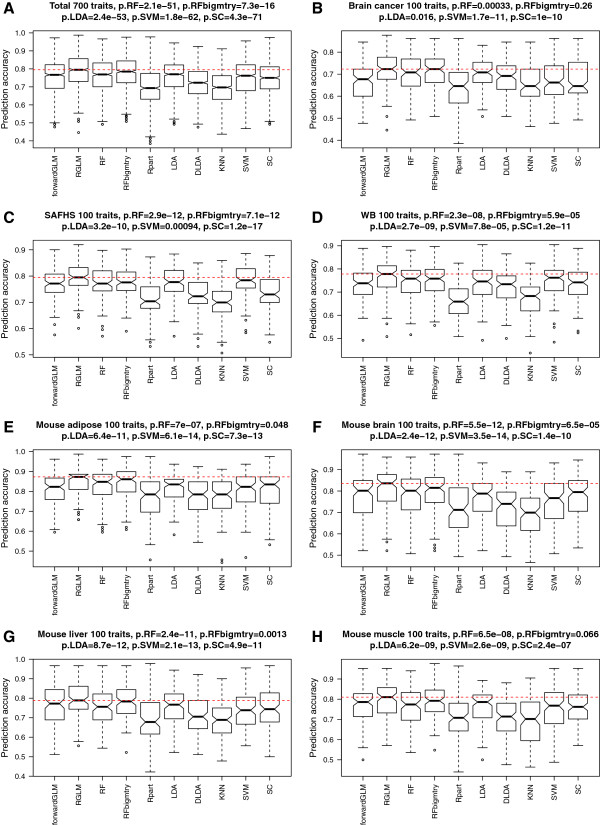
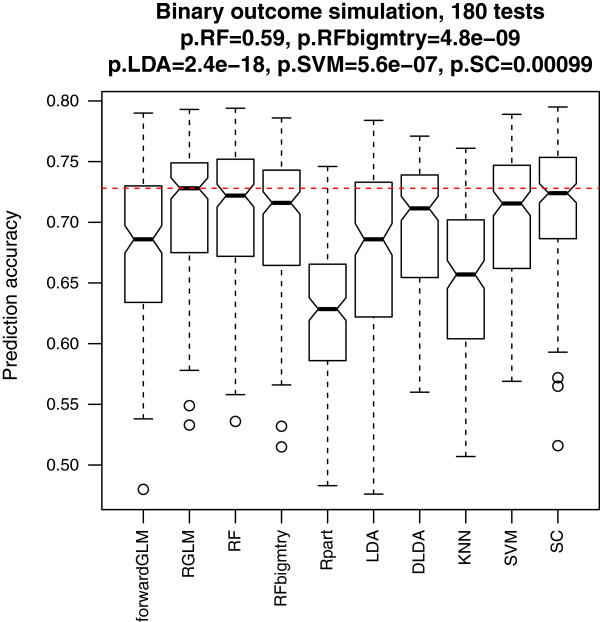
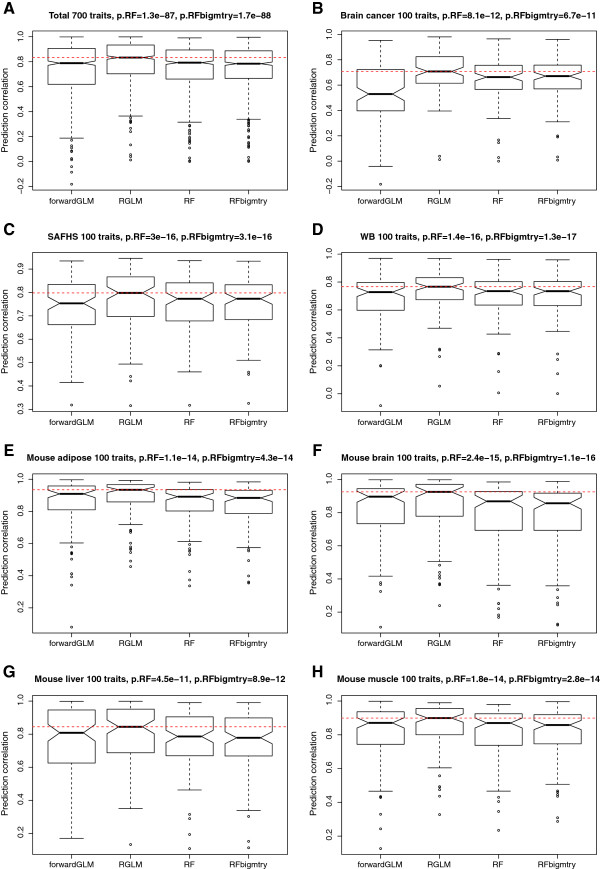
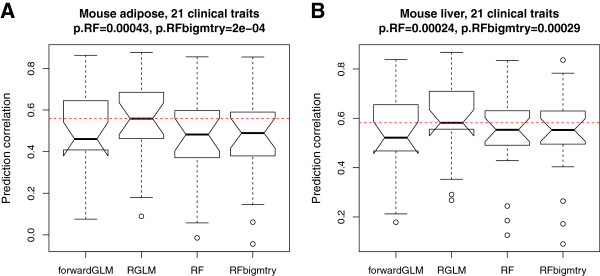
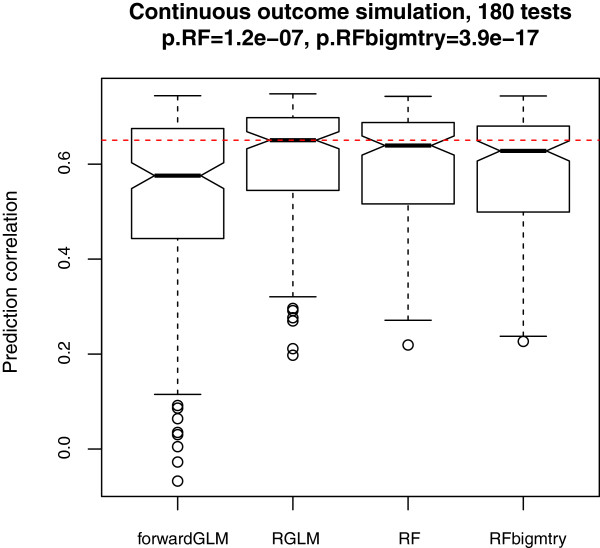
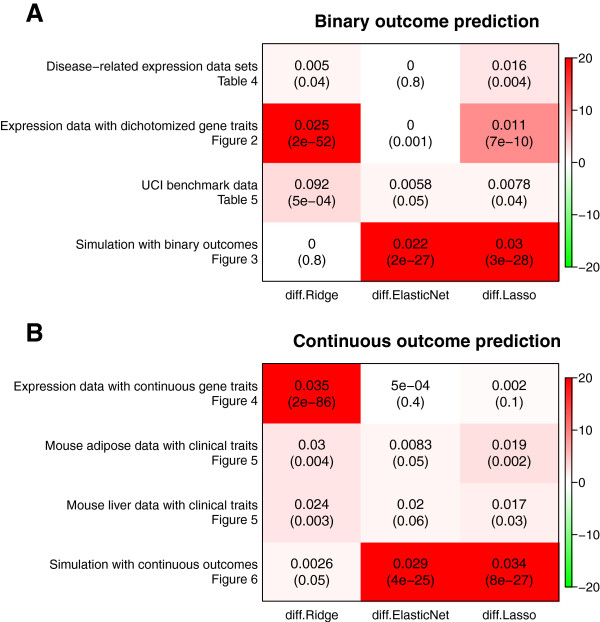
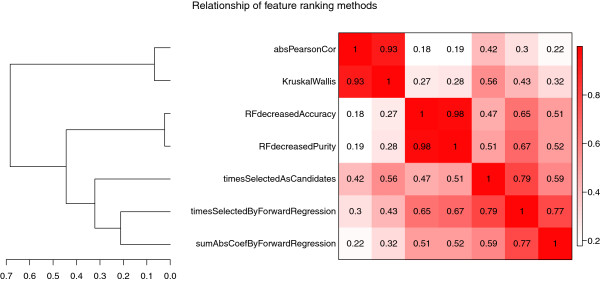
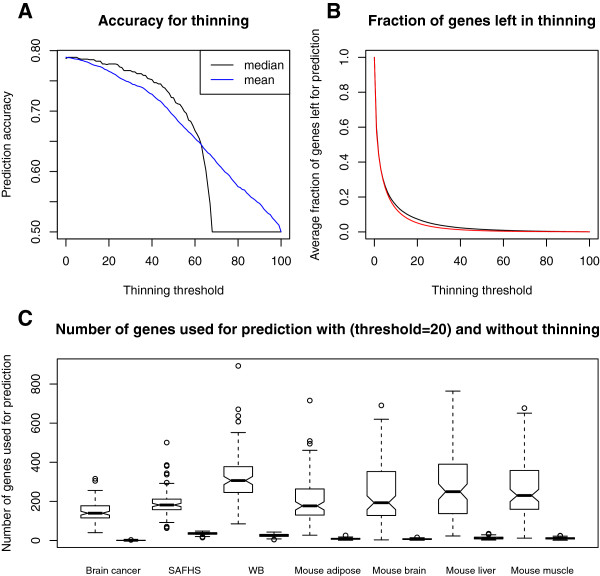
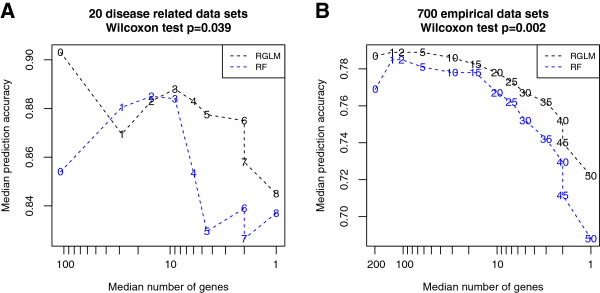
Results: Comprehensive evaluations involving hundreds of genomic data sets, the UCI machine learning benchmark data, and simulations are used to give GLM based ensemble predictors a new and careful look. A novel bootstrap aggregated (bagged) GLM predictor that incorporates several elements of randomness and instability (random subspace method, optional interaction terms, forward variable selection) often outperforms a host of alternative prediction methods including random forests and penalized regression models (ridge regression, elastic net, lasso). This random generalized linear model (RGLM) predictor provides variable importance measures that can be used to define a "thinned" ensemble predictor (involving few features) that retains excellent predictive accuracy.
Conclusion: RGLM is a state of the art predictor that shares the advantages of a random forest (excellent predictive accuracy, feature importance measures, out-of-bag estimates of accuracy) with those of a forward selected generalized linear model (interpretability). These methods are implemented in the freely available R software package randomGLM.
Figures
References
-
- Vapnik V. The nature of statistical learning theory. New York: Springer; 2000.
-
- Breiman L, Friedman J, Stone C, Olshen R. Classification and regression trees. California: Wadsworth International Group; 1984.
-
- Dudoit S, Fridlyand J, Speed TP. Comparison of discrimination methods for the classification of tumors using gene expression data. J Am Stat Assoc. 2002;97(457):77–87. doi: 10.1198/016214502753479248. - DOI
-
- Diaz-Uriarte R, Alvarez de AndresS. Gene selection and classification of microarray data using random forest. BMC Bioinformatics. 2006;7:3. doi: 10.1186/1471-2105-7-3. [ http://www.biomedcentral.com/1471-2105/7/3] - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
