

MotifLab: a tools and data integration workbench for motif discovery and regulatory sequence analysis
- PMID: 23323883
- PMCID: PMC3556059
- DOI: 10.1186/1471-2105-14-9
MotifLab: a tools and data integration workbench for motif discovery and regulatory sequence analysis
Abstract
Background: Traditional methods for computational motif discovery often suffer from poor performance. In particular, methods that search for sequence matches to known binding motifs tend to predict many non-functional binding sites because they fail to take into consideration the biological state of the cell. In recent years, genome-wide studies have generated a lot of data that has the potential to improve our ability to identify functional motifs and binding sites, such as information about chromatin accessibility and epigenetic states in different cell types. However, it is not always trivial to make use of this data in combination with existing motif discovery tools, especially for researchers who are not skilled in bioinformatics programming.
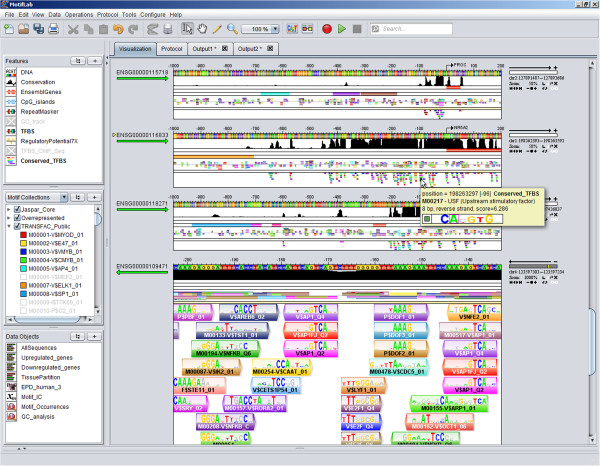
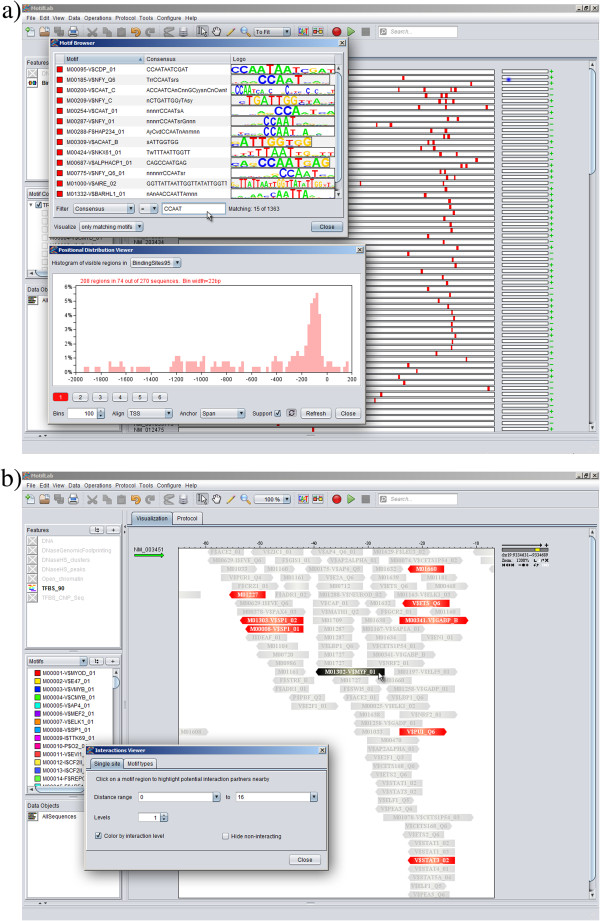
Results: Here we present MotifLab, a general workbench for analysing regulatory sequence regions and discovering transcription factor binding sites and cis-regulatory modules. MotifLab supports comprehensive motif discovery and analysis by allowing users to integrate several popular motif discovery tools as well as different kinds of additional information, including phylogenetic conservation, epigenetic marks, DNase hypersensitive sites, ChIP-Seq data, positional binding preferences of transcription factors, transcription factor interactions and gene expression. MotifLab offers several data-processing operations that can be used to create, manipulate and analyse data objects, and complete analysis workflows can be constructed and automatically executed within MotifLab, including graphical presentation of the results.
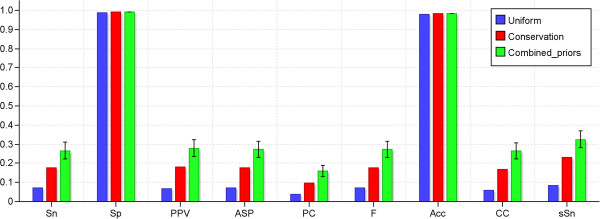
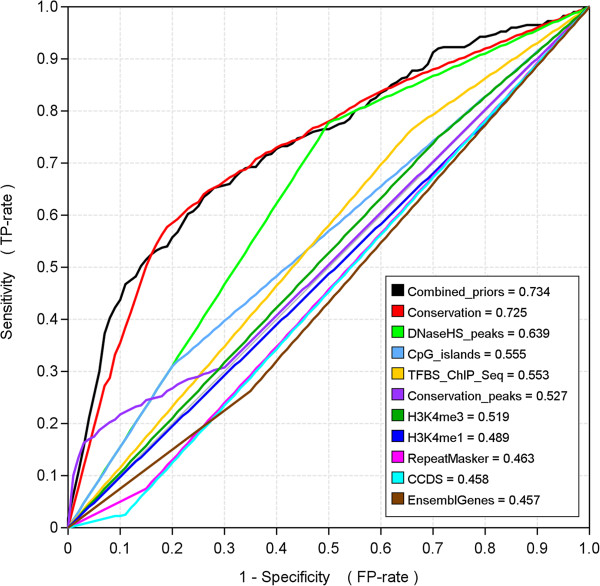
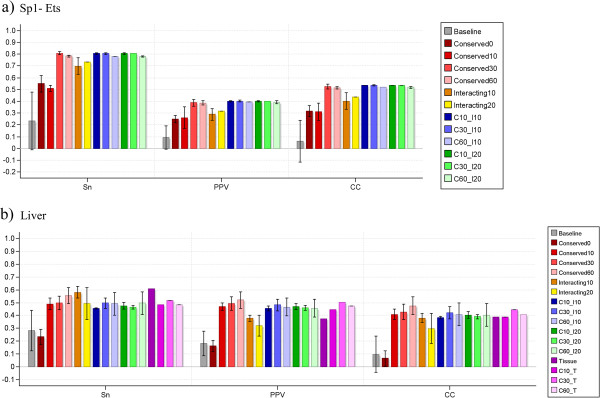
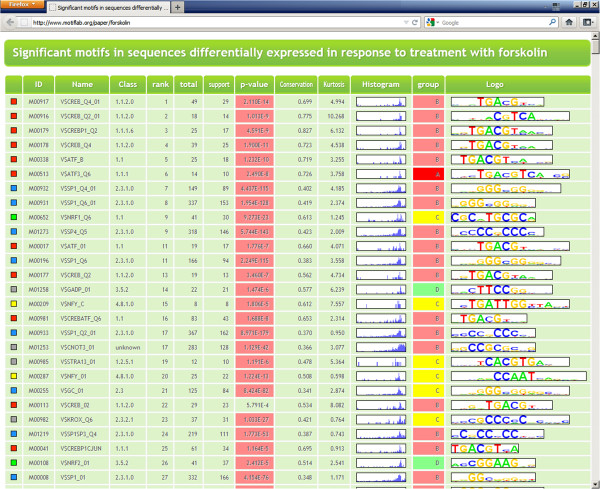
Conclusions: We have developed MotifLab as a flexible workbench for motif analysis in a genomic context. The flexibility and effectiveness of this workbench has been demonstrated on selected test cases, in particular two previously published benchmark data sets for single motifs and modules, and a realistic example of genes responding to treatment with forskolin. MotifLab is freely available at http://www.motiflab.org.
Figures
Similar articles
-
TrawlerWeb: an online de novo motif discovery tool for next-generation sequencing datasets.BMC Genomics. 2018 Apr 5;19(1):238. doi: 10.1186/s12864-018-4630-0. BMC Genomics. 2018. PMID: 29621972 Free PMC article.
-
Comparative analysis of regulatory motif discovery tools for transcription factor binding sites.Genomics Proteomics Bioinformatics. 2007 May;5(2):131-42. doi: 10.1016/S1672-0229(07)60023-0. Genomics Proteomics Bioinformatics. 2007. PMID: 17893078 Free PMC article.
-
Assessment of composite motif discovery methods.BMC Bioinformatics. 2008 Feb 26;9:123. doi: 10.1186/1471-2105-9-123. BMC Bioinformatics. 2008. PMID: 18302777 Free PMC article.
-
An algorithmic perspective of de novo cis-regulatory motif finding based on ChIP-seq data.Brief Bioinform. 2018 Sep 28;19(5):1069-1081. doi: 10.1093/bib/bbx026. Brief Bioinform. 2018. PMID: 28334268 Review.
-
Role of ChIP-seq in the discovery of transcription factor binding sites, differential gene regulation mechanism, epigenetic marks and beyond.Cell Cycle. 2014;13(18):2847-52. doi: 10.4161/15384101.2014.949201. Cell Cycle. 2014. PMID: 25486472 Free PMC article. Review.
Cited by
-
Large-scale identification of gibberellin-related transcription factors defines group VII ETHYLENE RESPONSE FACTORS as functional DELLA partners.Plant Physiol. 2014 Oct;166(2):1022-32. doi: 10.1104/pp.114.244723. Epub 2014 Aug 12. Plant Physiol. 2014. PMID: 25118255 Free PMC article.
-
The catecholamine biosynthetic enzyme dopamine β-hydroxylase (DBH): first genome-wide search positions trait-determining variants acting additively in the proximal promoter.Hum Mol Genet. 2014 Dec 1;23(23):6375-84. doi: 10.1093/hmg/ddu332. Epub 2014 Jun 30. Hum Mol Genet. 2014. PMID: 24986918 Free PMC article.
-
MODSIDE: a motif discovery pipeline and similarity detector.BMC Genomics. 2018 Oct 19;19(1):755. doi: 10.1186/s12864-018-5148-1. BMC Genomics. 2018. PMID: 30340511 Free PMC article.
-
DynaMIT: the dynamic motif integration toolkit.Nucleic Acids Res. 2016 Jan 8;44(1):e2. doi: 10.1093/nar/gkv807. Epub 2015 Aug 7. Nucleic Acids Res. 2016. PMID: 26253738 Free PMC article.
-
Genome Wide Binding Site Analysis Reveals Transcriptional Coactivation of Cytokinin-Responsive Genes by DELLA Proteins.PLoS Genet. 2015 Jul 2;11(7):e1005337. doi: 10.1371/journal.pgen.1005337. eCollection 2015 Jul. PLoS Genet. 2015. PMID: 26134422 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
