

xMSanalyzer: automated pipeline for improved feature detection and downstream analysis of large-scale, non-targeted metabolomics data
- PMID: 23323971
- PMCID: PMC3562220
- DOI: 10.1186/1471-2105-14-15
xMSanalyzer: automated pipeline for improved feature detection and downstream analysis of large-scale, non-targeted metabolomics data
Abstract
Background: Detection of low abundance metabolites is important for de novo mapping of metabolic pathways related to diet, microbiome or environmental exposures. Multiple algorithms are available to extract m/z features from liquid chromatography-mass spectral data in a conservative manner, which tends to preclude detection of low abundance chemicals and chemicals found in small subsets of samples. The present study provides software to enhance such algorithms for feature detection, quality assessment, and annotation.
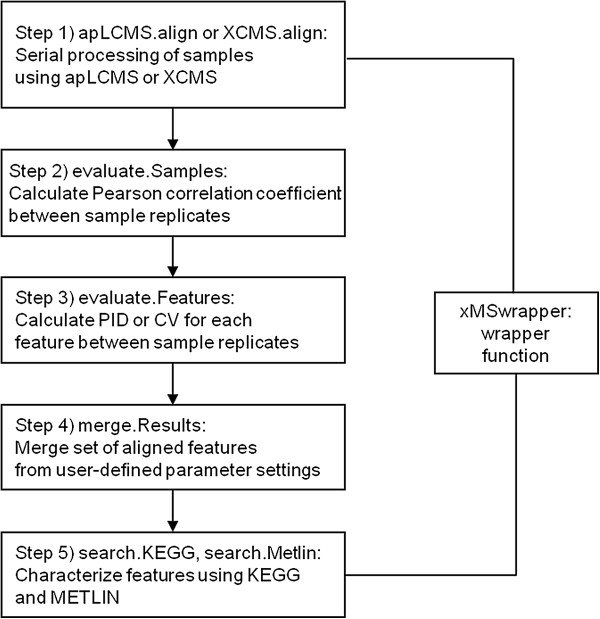
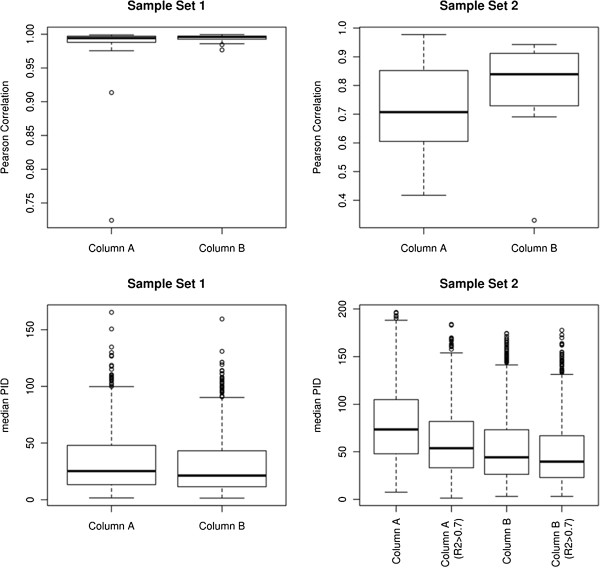
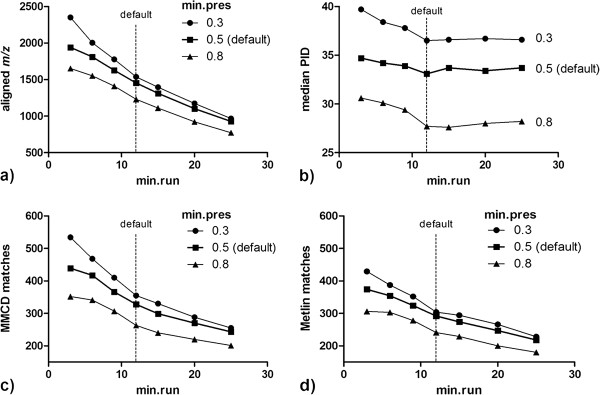
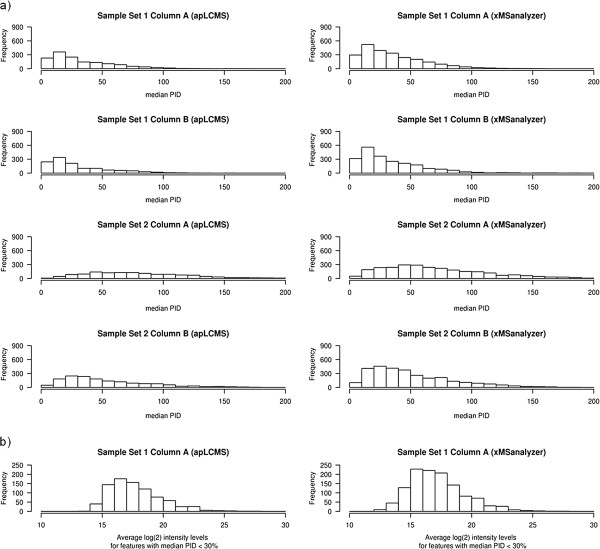
Results: xMSanalyzer is a set of utilities for automated processing of metabolomics data. The utilites can be classified into four main modules to: 1) improve feature detection for replicate analyses by systematic re-extraction with multiple parameter settings and data merger to optimize the balance between sensitivity and reliability, 2) evaluate sample quality and feature consistency, 3) detect feature overlap between datasets, and 4) characterize high-resolution m/z matches to small molecule metabolites and biological pathways using multiple chemical databases. The package was tested with plasma samples and shown to more than double the number of features extracted while improving quantitative reliability of detection. MS/MS analysis of a random subset of peaks that were exclusively detected using xMSanalyzer confirmed that the optimization scheme improves detection of real metabolites.
Conclusions: xMSanalyzer is a package of utilities for data extraction, quality control assessment, detection of overlapping and unique metabolites in multiple datasets, and batch annotation of metabolites. The program was designed to integrate with existing packages such as apLCMS and XCMS, but the framework can also be used to enhance data extraction for other LC/MS data software.
Figures
References
-
- Zhang H, Wang Y, Gu X, Zhou J, Yan C. Metabolomic profiling of human plasma in pancreatic cancer using pressurized capillary electrochromatography. Electrophoresis. 2011;32(3–4):340–347. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
