

Dynamic DNA methylation across diverse human cell lines and tissues
- PMID: 23325432
- PMCID: PMC3589544
- DOI: 10.1101/gr.147942.112
Dynamic DNA methylation across diverse human cell lines and tissues
Abstract
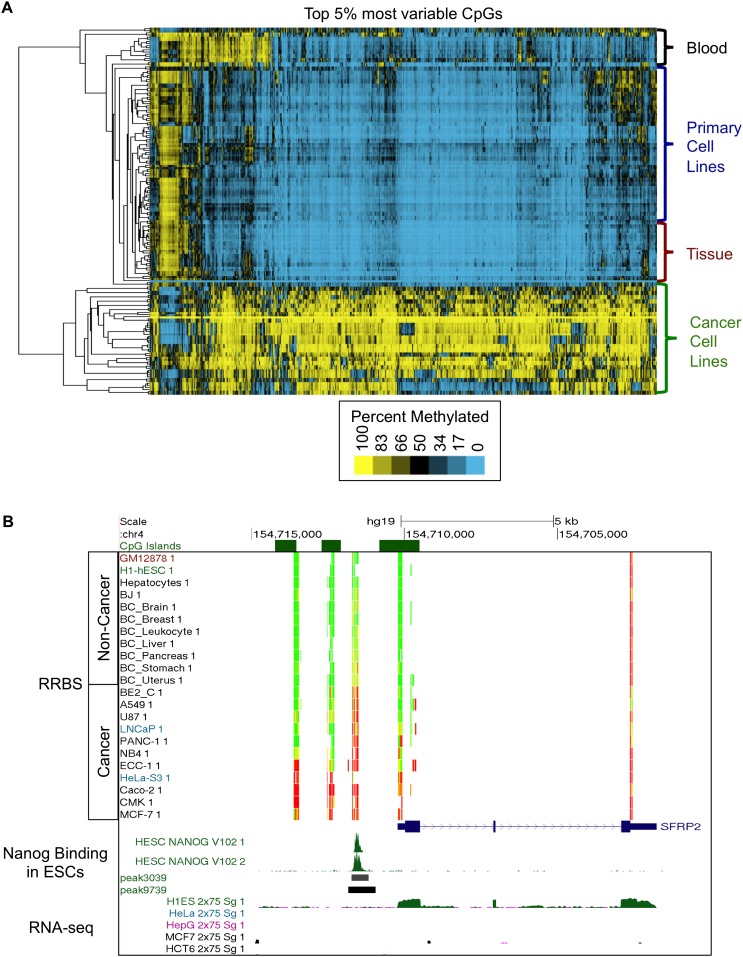
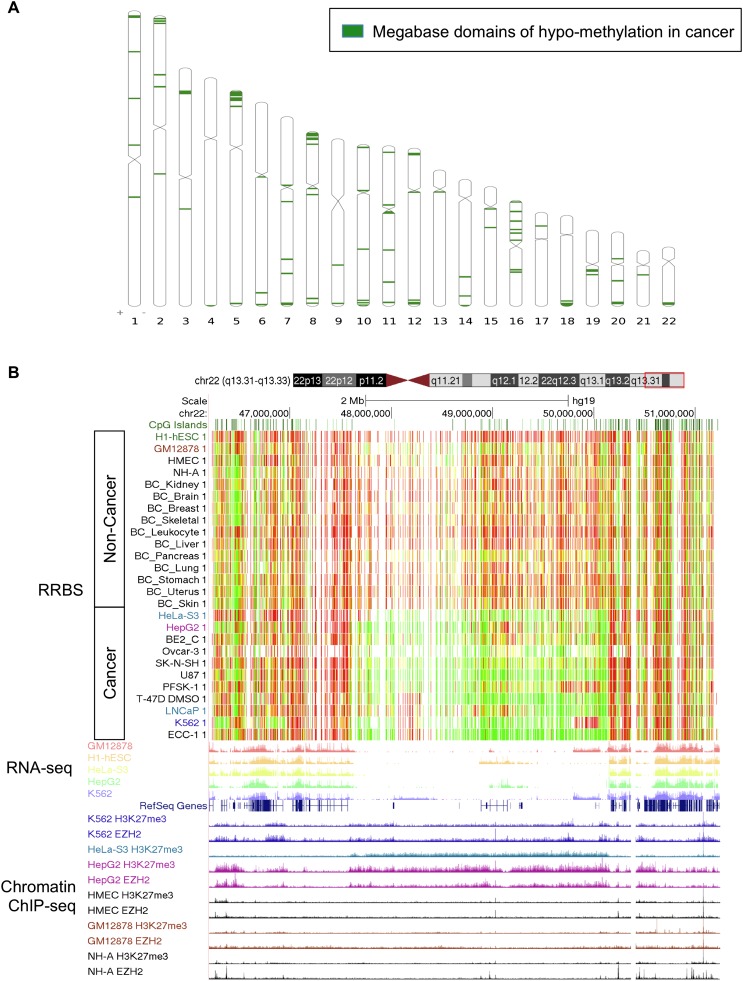
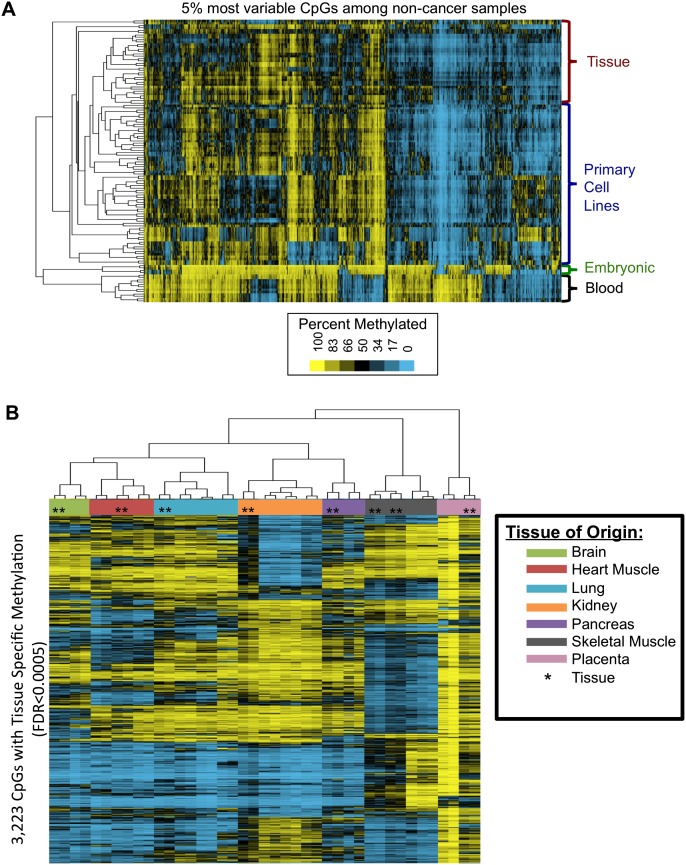
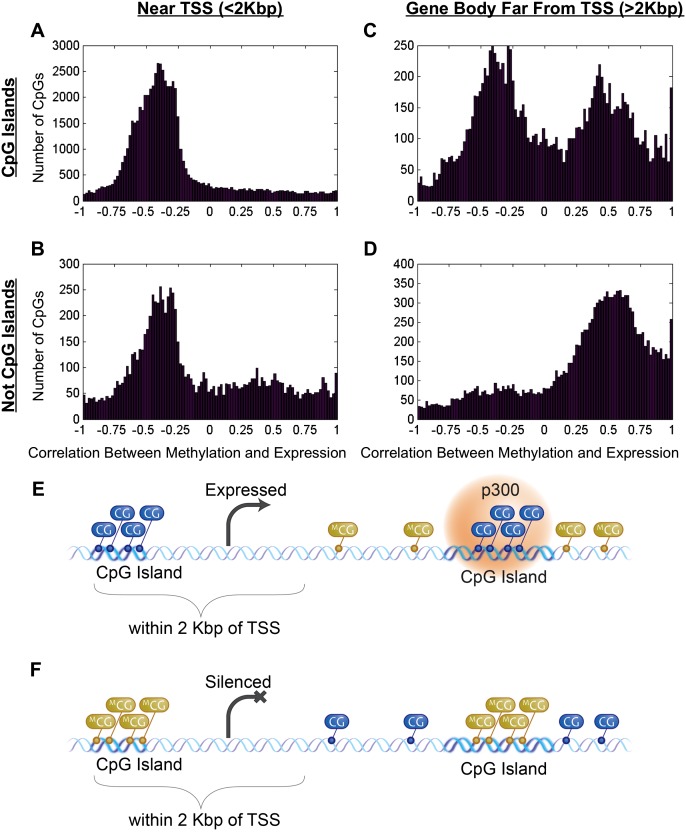
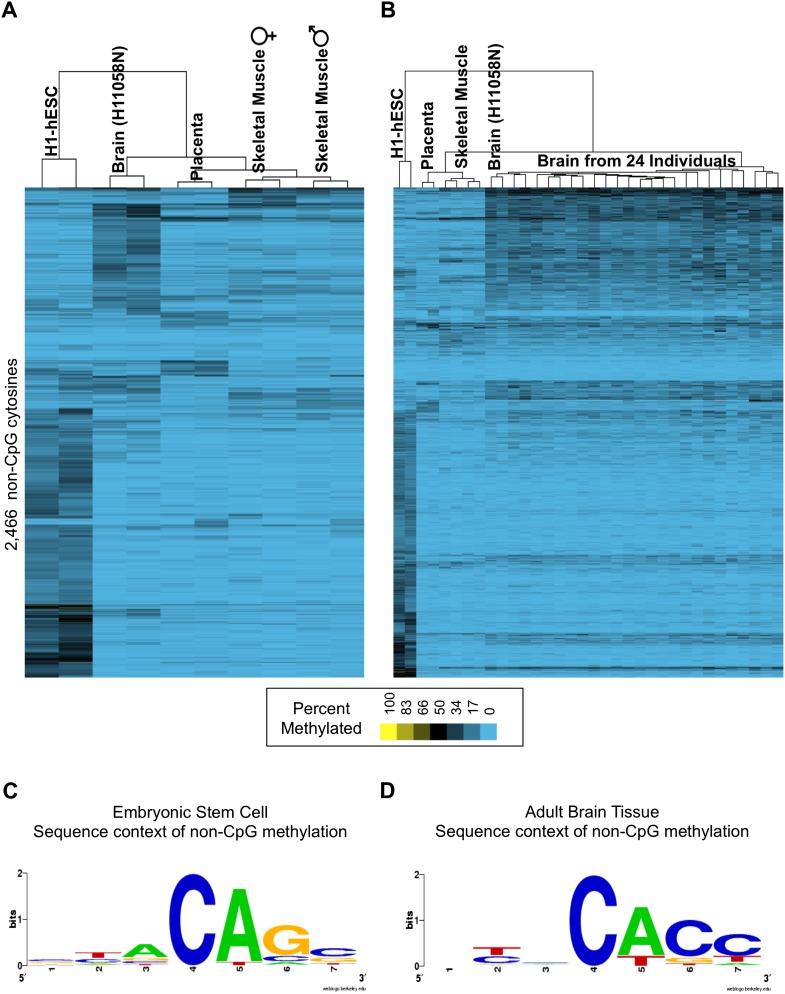
As studies of DNA methylation increase in scope, it has become evident that methylation has a complex relationship with gene expression, plays an important role in defining cell types, and is disrupted in many diseases. We describe large-scale single-base resolution DNA methylation profiling on a diverse collection of 82 human cell lines and tissues using reduced representation bisulfite sequencing (RRBS). Analysis integrating RNA-seq and ChIP-seq data illuminates the functional role of this dynamic mark. Loci that are hypermethylated across cancer types are enriched for sites bound by NANOG in embryonic stem cells, which supports and expands the model of a stem/progenitor cell signature in cancer. CpGs that are hypomethylated across cancer types are concentrated in megabase-scale domains that occur near the telomeres and centromeres of chromosomes, are depleted of genes, and are enriched for cancer-specific EZH2 binding and H3K27me3 (repressive chromatin). In noncancer samples, there are cell-type specific methylation signatures preserved in primary cell lines and tissues as well as methylation differences induced by cell culture. The relationship between methylation and expression is context-dependent, and we find that CpG-rich enhancers bound by EP300 in the bodies of expressed genes are unmethylated despite the dense gene-body methylation surrounding them. Non-CpG cytosine methylation occurs in human somatic tissue, is particularly prevalent in brain tissue, and is reproducible across many individuals. This study provides an atlas of DNA methylation across diverse and well-characterized samples and enables new discoveries about DNA methylation and its role in gene regulation and disease.
Figures
References
-
- Aran D, Toperoff G, Rosenberg M, Hellman A 2011. Replication timing-related and gene body-specific methylation of active human genes. Hum Mol Genet 20: 670–680 - PubMed
-
- Berman BP, Weisenberger DJ, Aman JF, Hinoue T, Ramjan Z, Liu Y, Noushmehr H, Lange CP, van Dijk CM, Tollenaar RA, et al. 2012. Regions of focal DNA hypermethylation and long-range hypomethylation in colorectal cancer coincide with nuclear lamina-associated domains. Nat Genet 44: 40–46 - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous