

Sparsely correlated hidden Markov models with application to genome-wide location studies
- PMID: 23325620
- PMCID: PMC3582268
- DOI: 10.1093/bioinformatics/btt012
Sparsely correlated hidden Markov models with application to genome-wide location studies
Abstract
Motivation: Multiply correlated datasets have become increasingly common in genome-wide location analysis of regulatory proteins and epigenetic modifications. Their correlation can be directly incorporated into a statistical model to capture underlying biological interactions, but such modeling quickly becomes computationally intractable.
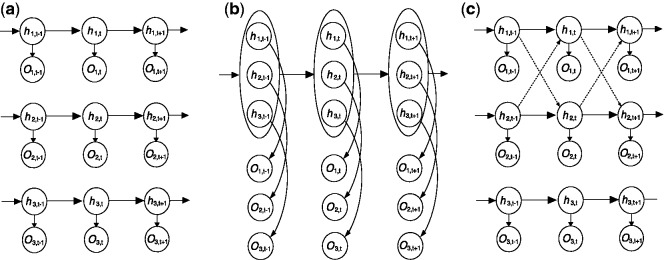
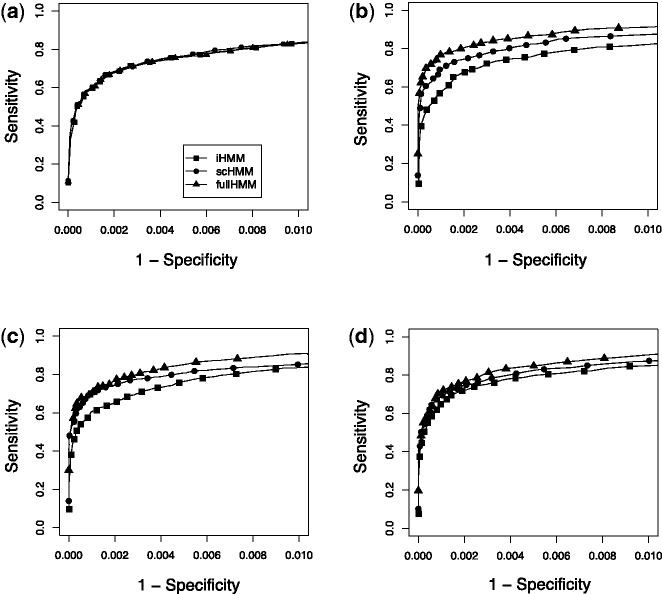
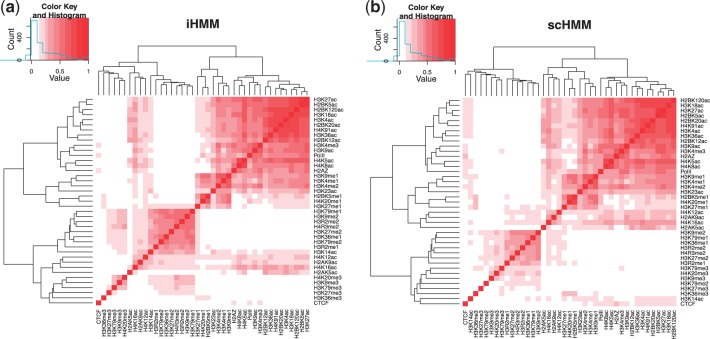
Results: We present sparsely correlated hidden Markov models (scHMM), a novel method for performing simultaneous hidden Markov model (HMM) inference for multiple genomic datasets. In scHMM, a single HMM is assumed for each series, but the transition probability in each series depends on not only its own hidden states but also the hidden states of other related series. For each series, scHMM uses penalized regression to select a subset of the other data series and estimate their effects on the odds of each transition in the given series. Following this, hidden states are inferred using a standard forward-backward algorithm, with the transition probabilities adjusted by the model at each position, which helps retain the order of computation close to fitting independent HMMs (iHMM). Hence, scHMM is a collection of inter-dependent non-homogeneous HMMs, capable of giving a close approximation to a fully multivariate HMM fit. A simulation study shows that scHMM achieves comparable sensitivity to the multivariate HMM fit at a much lower computational cost. The method was demonstrated in the joint analysis of 39 histone modifications, CTCF and RNA polymerase II in human CD4+ T cells. scHMM reported fewer high-confidence regions than iHMM in this dataset, but scHMM could recover previously characterized histone modifications in relevant genomic regions better than iHMM. In addition, the resulting combinatorial patterns from scHMM could be better mapped to the 51 states reported by the multivariate HMM method of Ernst and Kellis.
Availability: The scHMM package can be freely downloaded from http://sourceforge.net/p/schmm/ and is recommended for use in a linux environment.
Figures
Similar articles
-
Computationally Tractable Multivariate HMM in Genome-Wide Mapping Studies.Methods Mol Biol. 2017;1552:135-148. doi: 10.1007/978-1-4939-6753-7_10. Methods Mol Biol. 2017. PMID: 28224496
-
Annotation of genomics data using bidirectional hidden Markov models unveils variations in Pol II transcription cycle.Mol Syst Biol. 2014 Dec 19;10(12):768. doi: 10.15252/msb.20145654. Mol Syst Biol. 2014. PMID: 25527639 Free PMC article.
-
Hidden Markov Models in Bioinformatics: SNV Inference from Next Generation Sequence.Methods Mol Biol. 2017;1552:123-133. doi: 10.1007/978-1-4939-6753-7_9. Methods Mol Biol. 2017. PMID: 28224495
-
An Introduction to Infinite HMMs for Single-Molecule Data Analysis.Biophys J. 2017 May 23;112(10):2021-2029. doi: 10.1016/j.bpj.2017.04.027. Biophys J. 2017. PMID: 28538142 Free PMC article. Review.
-
Hidden Markov model and its applications in motif findings.Methods Mol Biol. 2010;620:405-16. doi: 10.1007/978-1-60761-580-4_13. Methods Mol Biol. 2010. PMID: 20652513 Review.
Cited by
-
Chromatin-state discovery and genome annotation with ChromHMM.Nat Protoc. 2017 Dec;12(12):2478-2492. doi: 10.1038/nprot.2017.124. Epub 2017 Nov 9. Nat Protoc. 2017. PMID: 29120462 Free PMC article. Review.
-
Joint analysis of expression profiles from multiple cancers improves the identification of microRNA-gene interactions.Bioinformatics. 2013 Sep 1;29(17):2137-45. doi: 10.1093/bioinformatics/btt341. Epub 2013 Jun 14. Bioinformatics. 2013. PMID: 23772050 Free PMC article.
-
Bayesian adaptive group lasso with semiparametric hidden Markov models.Stat Med. 2019 Apr 30;38(9):1634-1650. doi: 10.1002/sim.8051. Epub 2018 Nov 28. Stat Med. 2019. PMID: 30484887 Free PMC article.
-
Integrating Epigenomics into the Understanding of Biomedical Insight.Bioinform Biol Insights. 2016 Dec 4;10:267-289. doi: 10.4137/BBI.S38427. eCollection 2016. Bioinform Biol Insights. 2016. PMID: 27980397 Free PMC article. Review.
References
-
- Bannister A, et al. Spatial distribution of di- and tri-methyl lysine 36 of histone H3 at active genes. J. Biol. Chem. 2005;280:17732–17736. - PubMed
-
- Barski A, et al. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. - PubMed
-
- Bernstein B, et al. The mammalian epigenome. Cell. 2007;128:669–681. - PubMed
-
- Churchill G. Stochastic models for heterogeneous DNA sequences. Bull. Math. Biol. 1989;51:79–94. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
