

Aberration in DNA methylation in B-cell lymphomas has a complex origin and increases with disease severity
- PMID: 23326238
- PMCID: PMC3542081
- DOI: 10.1371/journal.pgen.1003137
Aberration in DNA methylation in B-cell lymphomas has a complex origin and increases with disease severity
Abstract
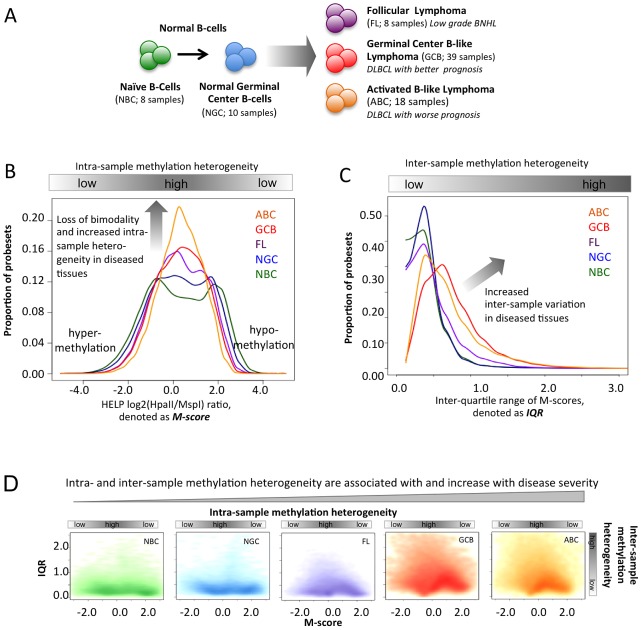
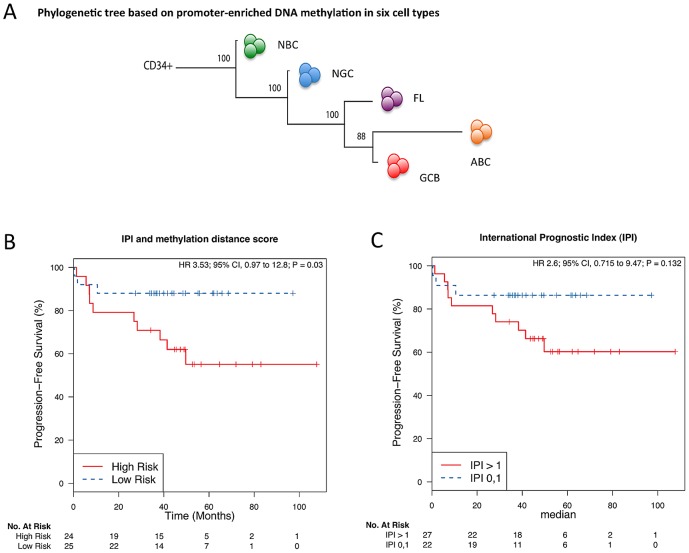
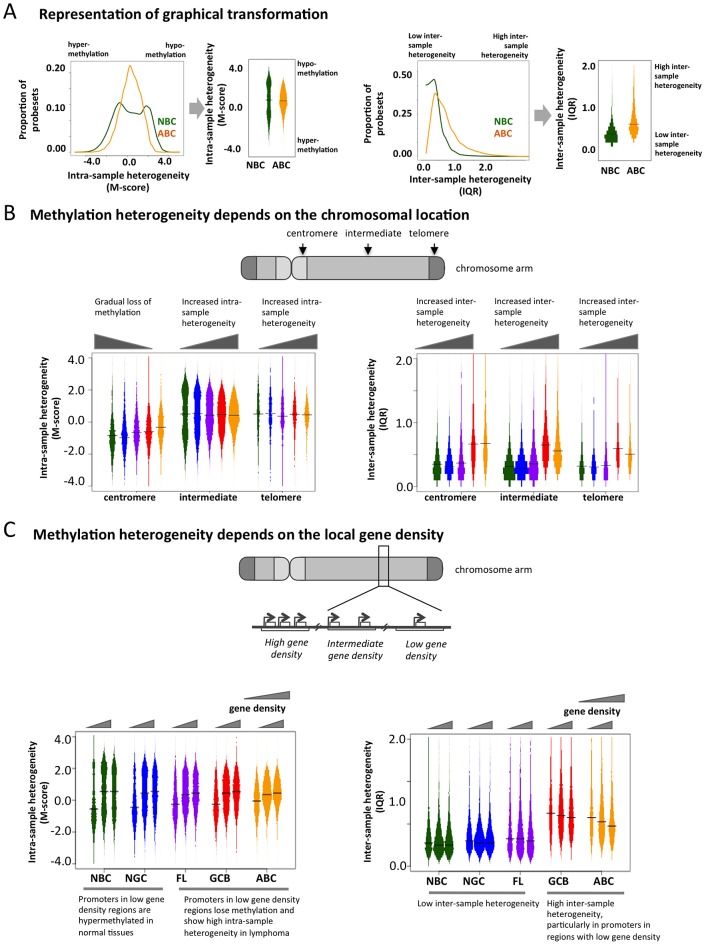
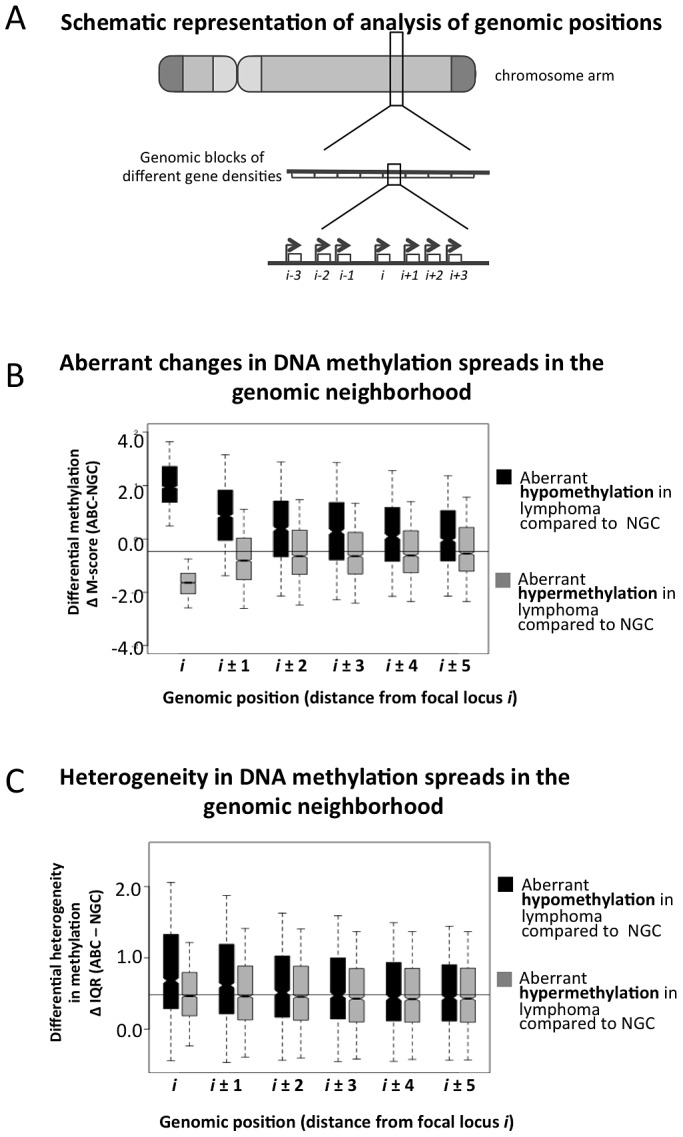
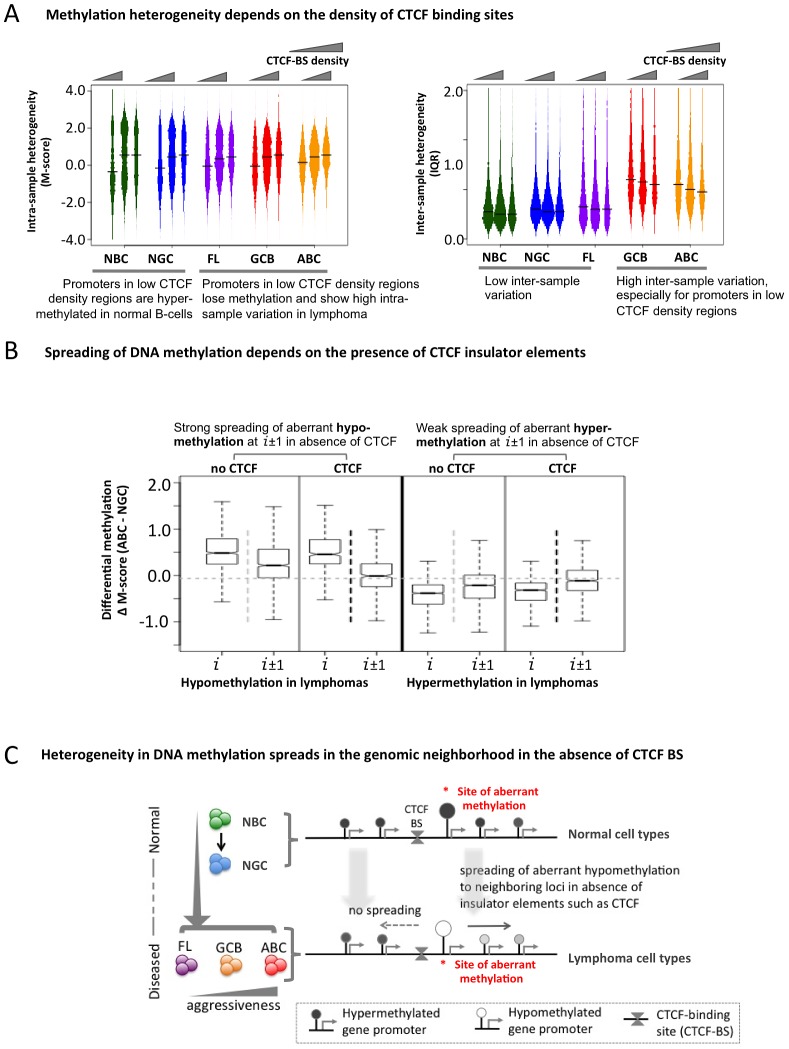
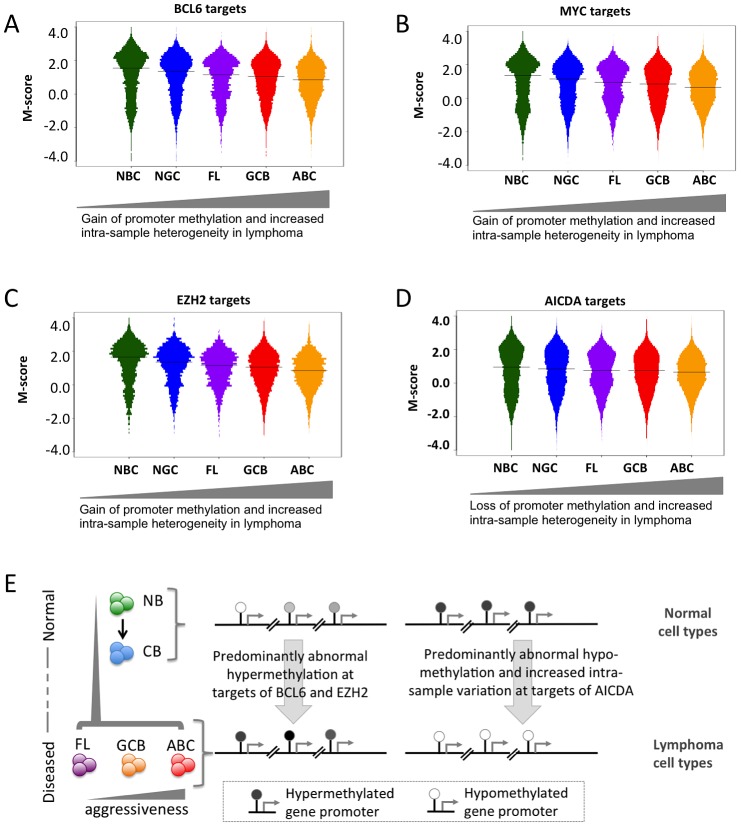
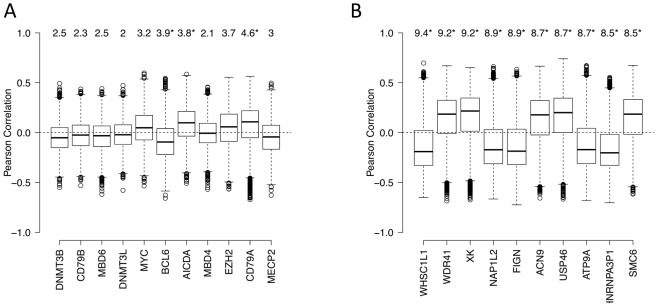
Despite mounting evidence that epigenetic abnormalities play a key role in cancer biology, their contributions to the malignant phenotype remain poorly understood. Here we studied genome-wide DNA methylation in normal B-cell populations and subtypes of B-cell non-Hodgkin lymphoma: follicular lymphoma and diffuse large B-cell lymphomas. These lymphomas display striking and progressive intra-tumor heterogeneity and also inter-patient heterogeneity in their cytosine methylation patterns. Epigenetic heterogeneity is initiated in normal germinal center B-cells, increases markedly with disease aggressiveness, and is associated with unfavorable clinical outcome. Moreover, patterns of abnormal methylation vary depending upon chromosomal regions, gene density and the status of neighboring genes. DNA methylation abnormalities arise via two distinct processes: i) lymphomagenic transcriptional regulators perturb promoter DNA methylation in a target gene-specific manner, and ii) aberrant epigenetic states tend to spread to neighboring promoters in the absence of CTCF insulator binding sites.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- TNHLCP (1997) A clinical evaluation of the International Lymphoma Study Group classification of non-Hodgkin's lymphoma. The Non-Hodgkin's Lymphoma Classification Project. Blood 89: 3909–3918. - PubMed
-
- Tan D, Horning SJ (2008) Follicular lymphoma: clinical features and treatment. Hematol Oncol Clin North Am 22: 863–882, viii. - PubMed
-
- Rosenwald A, Wright G, Chan WC, Connors JM, Campo E, et al. (2002) The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N Engl J Med 346: 1937–1947. - PubMed
-
- Klein U, Dalla-Favera R (2008) Germinal centres: role in B-cell physiology and malignancy. Nat Rev Immunol 8: 22–33. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
