

Chronic nicotine exposure augments renal oxidative stress and injury through transcriptional activation of p66shc
- PMID: 23328708
- PMCID: PMC3685305
- DOI: 10.1093/ndt/gfs596
Chronic nicotine exposure augments renal oxidative stress and injury through transcriptional activation of p66shc
Abstract
Background: Chronic nicotine (Ch-NIC) exposure exacerbates ischemia/reperfusion (I/R)-induced oxidative stress and acute kidney injury (AKI), and mitochondrial production of reactive oxygen species (ROS) in cultured renal proximal tubule cells (RPTCs). Because Ser36-phosphorylated p66shc modulates mitochondrial ROS production and injury of RPTCs, we hypothesized that Ch-NIC exacerbates AKI by increasing stress-induced phosphorylation of p66shc.
Methods: We first tested whether Ch-NIC augments I/R-AKI-induced expression and phosphorylation of p66shc in vivo. We then examined whether knocking down p66shc, or impairing its Ser36 phosphorylation or binding to cytochrome c, alters the effects of Ch-NIC on oxidative stress (H₂O₂)-induced production of ROS, mitochondrial depolarization and injury in RPTCs in vitro.
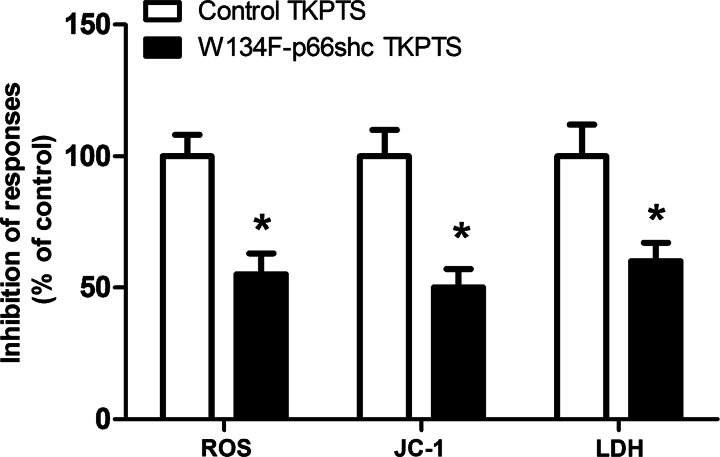
Results: We found that Ch-NIC increased the expression of p66shc in the control and ischemic kidneys, but only increased its Ser36 phosphorylation after renal I/R. Knocking down p66shc or impairing phosphorylation of its Ser36 residue, via the S36A mutation (but not the phosphomimetic S36D mutation), blunted Ch-NIC + H2O2-dependent ROS production, mitochondrial depolarization and injury in RPTCs. Additionally, Ch-NIC + H2O2-dependent binding of p66shc to mitochondrial cytochrome c was attenuated by S36A mutation of p66shc, and impairing cytochrome c binding (via W134F mutation) abolished ROS production, mitochondrial depolarization and injury, while ectopic overexpression of p66shc (which mimics Ch-NIC treatment) augmented oxidant injury. We determined that Ch-NIC stimulates the p66shc promoter through p53- and epigenetic modification (promoter hypomethylation).
Conclusions: Ch-NIC worsens oxidative stress-dependent acute renal injury by increasing expression and consequent oxidative stress-dependent Ser36 phosphorylation of p66shc. Thus, targeting this pathway may have therapeutic relevance in preventing/ameliorating tobacco-related kidney injury.
Keywords: ROS; cytochrome c; mitochondria; nicotine; p66shc.
Figures

Comment in
-
Chronic nicotine exposure and acute kidney injury: new concepts and experimental evidence.Nephrol Dial Transplant. 2013 Jun;28(6):1329-31. doi: 10.1093/ndt/gft019. Epub 2013 Feb 28. Nephrol Dial Transplant. 2013. PMID: 23449342 No abstract available.
References
-
- Orth SR, Ritz E, Schrier RW. The renal risks of smoking. Kidney Int. 1997;51:1669–1677. doi:10.1038/ki.1997.232. - DOI - PubMed
-
- Jaimes EA, Tian RX, Raij L. Nicotine: the link between cigarette smoking and the progression of renal injury? Am J Physiol Heart Circ Physiol. 2007;292:H76–H82. doi:10.1152/ajpheart.00693.2006. - DOI - PubMed
-
- Hua P, Feng W, Ji S, et al. Nicotine worsens the severity of nephropathy in diabetic mice: implications for the progression of kidney disease in smokers. Am J Physiol Renal Physiol. 2010;299:F732–F739. doi:10.1152/ajprenal.00293.2010. - DOI - PMC - PubMed
-
- Jaimes EA, Tian RX, Joshi MS, et al. Nicotine augments glomerular injury in a rat model of acute nephritis. Am J Nephrol. 2009;29:319–326. doi:10.1159/000163593. - DOI - PMC - PubMed
-
- Orth SR, Viedt C, Ritz E. Adverse effects of smoking in the renal patient. Tohoku J Exp Med. 2001;194:1–15. doi:10.1620/tjem.194.1. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
