

Nuclear-receptor-mediated regulation of drug- and bile-acid-transporter proteins in gut and liver
- PMID: 23330541
- PMCID: PMC4557796
- DOI: 10.3109/03602532.2012.748793
Nuclear-receptor-mediated regulation of drug- and bile-acid-transporter proteins in gut and liver
Abstract
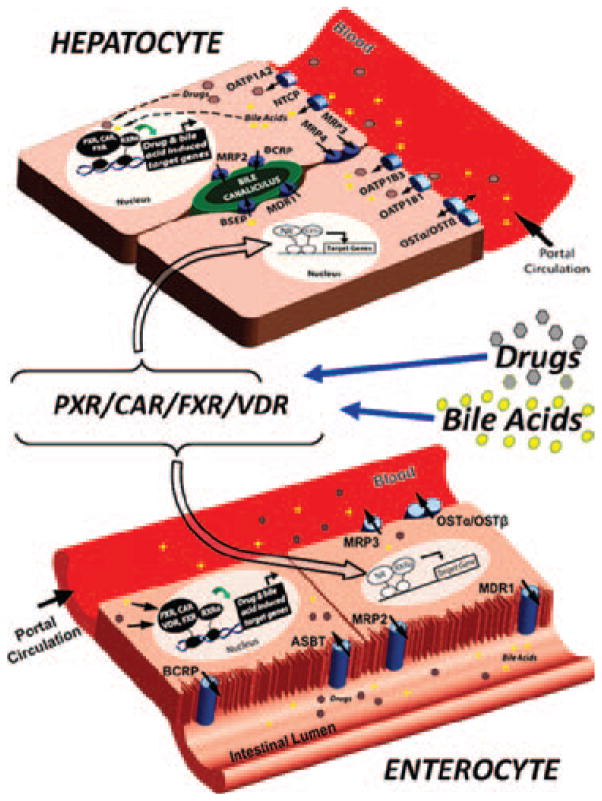
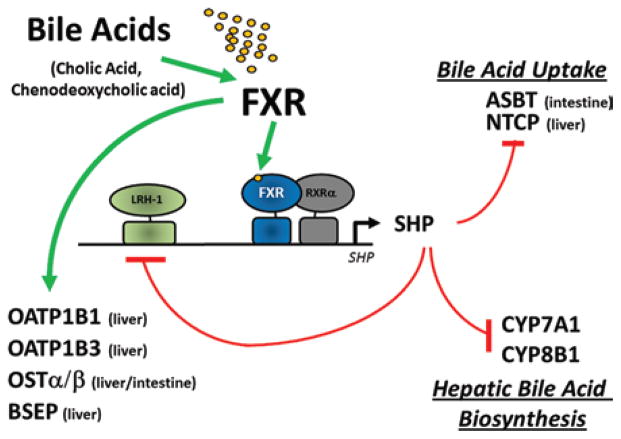
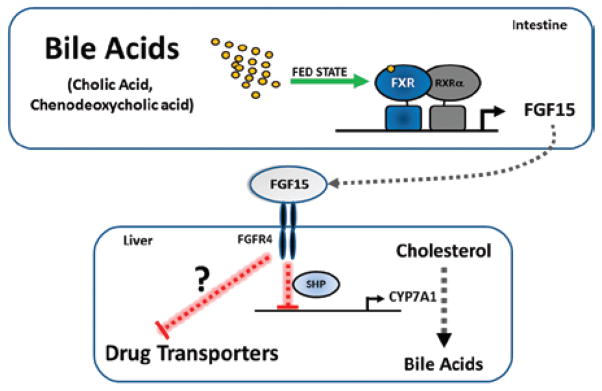
Adverse drug events (ADEs) are a common cause of patient morbidity and mortality and are classically thought to result, in part, from variation in expression and activity of hepatic enzymes of drug metabolism. It is now known that alterations in the expression of genes that encode drug- and bile-acid-transporter proteins in both the gut and liver play a previously unrecognized role in determining patient drug response and eventual clinical outcome. Four nuclear receptor (NR) superfamily members, including pregnane X receptor (PXR, NR1I2), constitutive androstane receptor (NR1I3), farnesoid X receptor (NR1H4), and vitamin D receptor (NR1I1), play pivotal roles in drug- and bile-acid-activated programs of gene expression to coordinately regulate drug- and bile-acid transport activity in the intestine and liver. This review focuses on the NR-mediated gene activation of drug and bile-acid transporters in these tissues as well as the possible underlying molecular mechanisms.
Conflict of interest statement
The authors declare no financial conflicts of interest. The authors alone are responsible for the content and writing of this paper.
Figures
References
-
- Aiba T, Susa M, Fukumori S, Hashimoto Y. The effects of culture conditions on CYP3A4 and MDR1 mRNA induction by 1alpha,25-dihydroxyvitamin D(3) in human intestinal cell lines, Caco-2 and LS180. Drug Metab Pharmacokinet. 2005;20:268–274. - PubMed
-
- Allen JD, Schinkel AH. Multidrug resistance and pharmacological protection mediated by the breast cancer resistance protein (BCRP/ABCG2) Mol Cancer Ther. 2002;1:427–434. - PubMed
-
- Ananthanarayanan M, Balasubramanian N, Makishima M, Mangelsdorf DJ, Suchy FJ. Human bile salt export pump promoter is transactivated by the farnesoid X receptor/bile acid receptor. J Biol Chem. 2001;276:28857–28865. - PubMed
-
- Assem M, Schuetz EG, Leggas M, Sun D, Yasuda K, Reid G, et al. Interactions between hepatic Mrp4 and Sult2a as revealed by the constitutive androstane receptor and Mrp4 knockout mice. J Biol Chem. 2004;279:22250–22257. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases