

Genome-wide chromatin state transitions associated with developmental and environmental cues
- PMID: 23333102
- PMCID: PMC3563935
- DOI: 10.1016/j.cell.2012.12.033
Genome-wide chromatin state transitions associated with developmental and environmental cues
Abstract
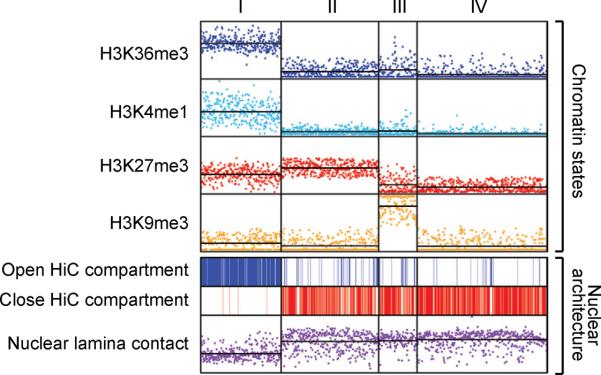
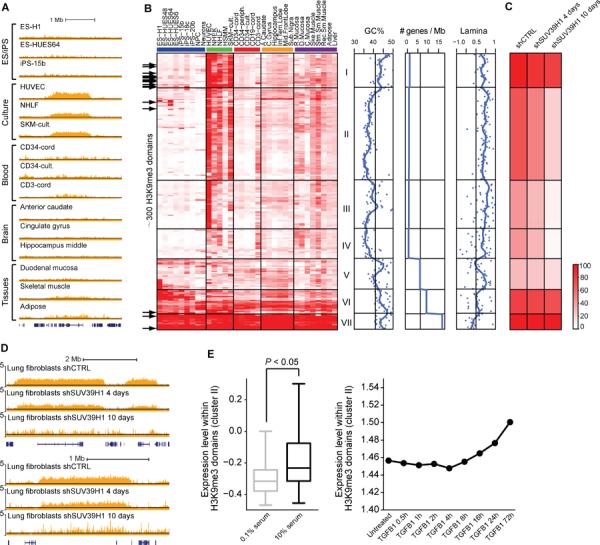
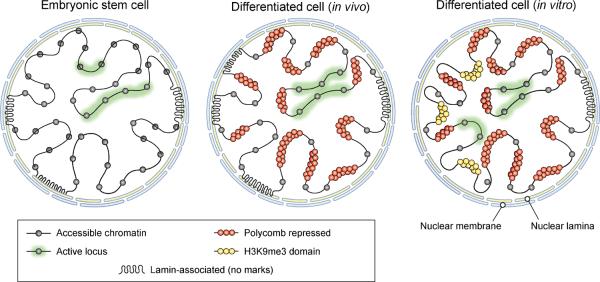
Differences in chromatin organization are key to the multiplicity of cell states that arise from a single genetic background, yet the landscapes of in vivo tissues remain largely uncharted. Here, we mapped chromatin genome-wide in a large and diverse collection of human tissues and stem cells. The maps yield unprecedented annotations of functional genomic elements and their regulation across developmental stages, lineages, and cellular environments. They also reveal global features of the epigenome, related to nuclear architecture, that also vary across cellular phenotypes. Specifically, developmental specification is accompanied by progressive chromatin restriction as the default state transitions from dynamic remodeling to generalized compaction. Exposure to serum in vitro triggers a distinct transition that involves de novo establishment of domains with features of constitutive heterochromatin. We describe how these global chromatin state transitions relate to chromosome and nuclear architecture, and discuss their implications for lineage fidelity, cellular senescence, and reprogramming.
Copyright © 2013 Elsevier Inc. All rights reserved.
Figures
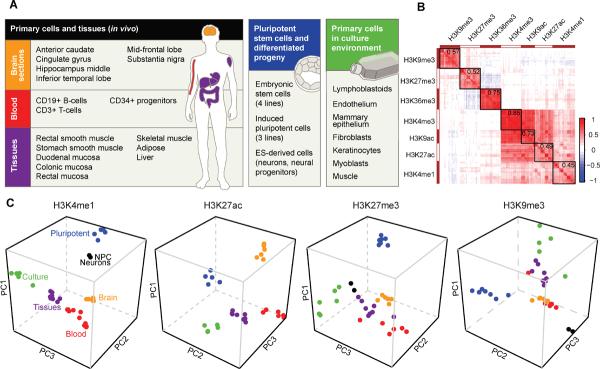
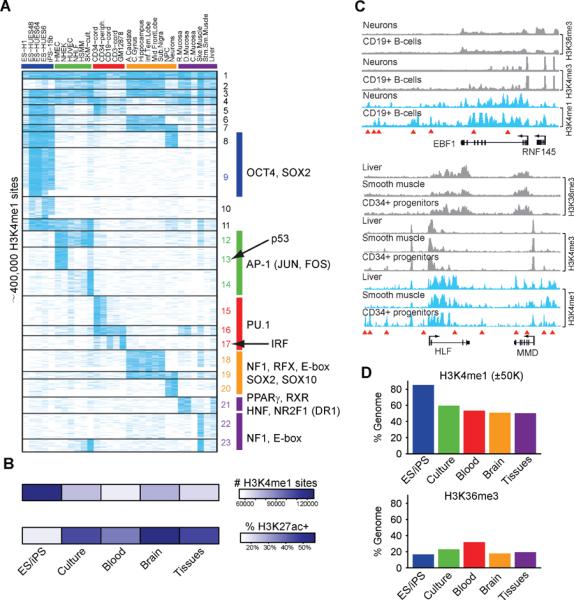
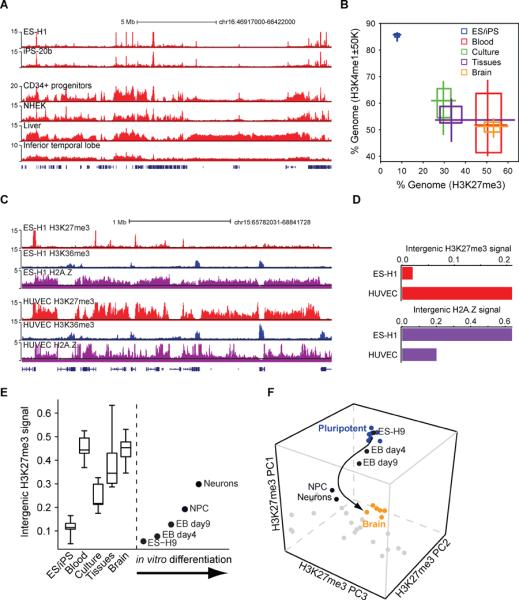
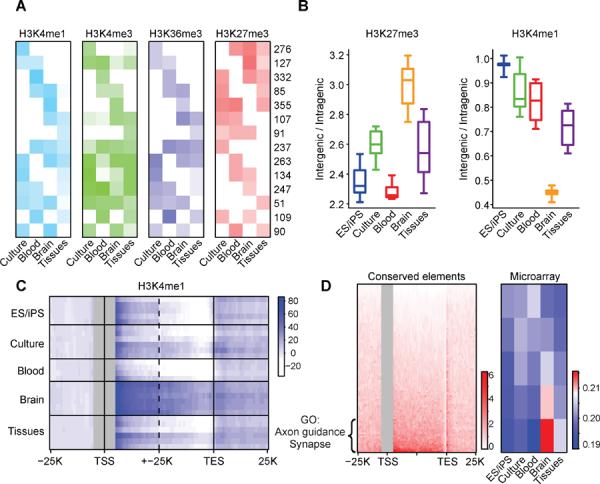
References
-
- Angel P, Karin M. The role of Jun, Fos and the AP-1 complex in cell-proliferation and transformation. Biochim Biophys Acta. 1991;1072:129–157. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
