

The mitotic kinase Aurora--a promotes distant metastases by inducing epithelial-to-mesenchymal transition in ERα(+) breast cancer cells
- PMID: 23334326
- PMCID: PMC4058768
- DOI: 10.1038/onc.2012.628
The mitotic kinase Aurora--a promotes distant metastases by inducing epithelial-to-mesenchymal transition in ERα(+) breast cancer cells
Abstract
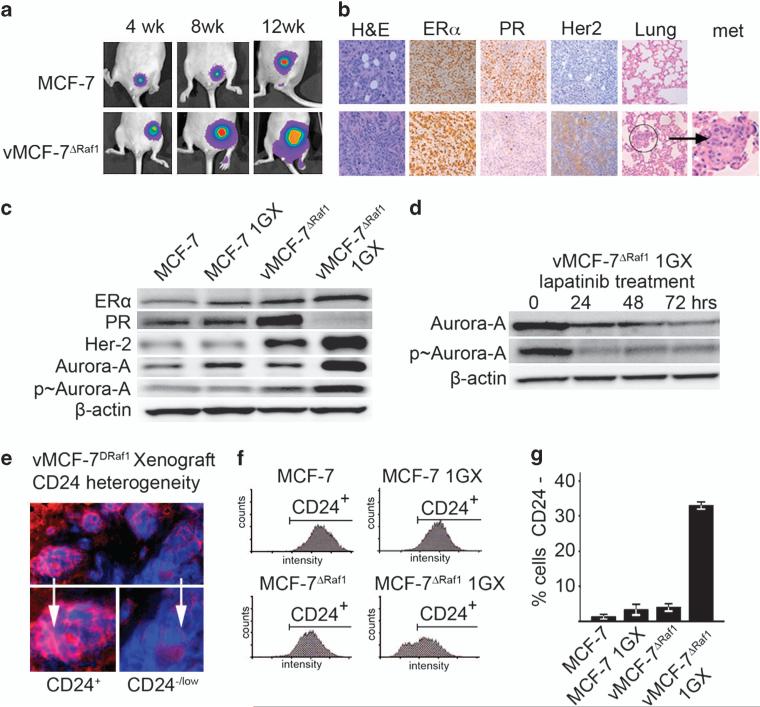
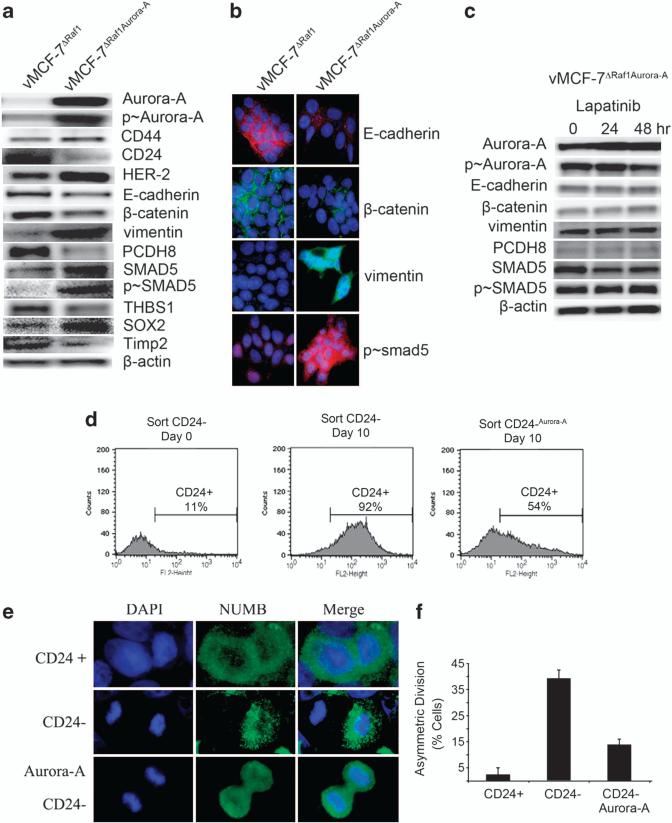
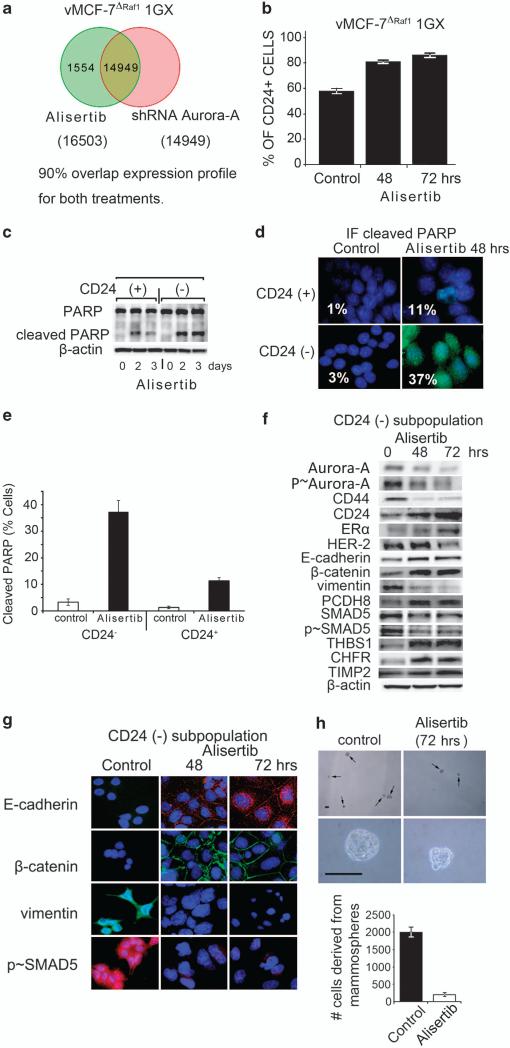
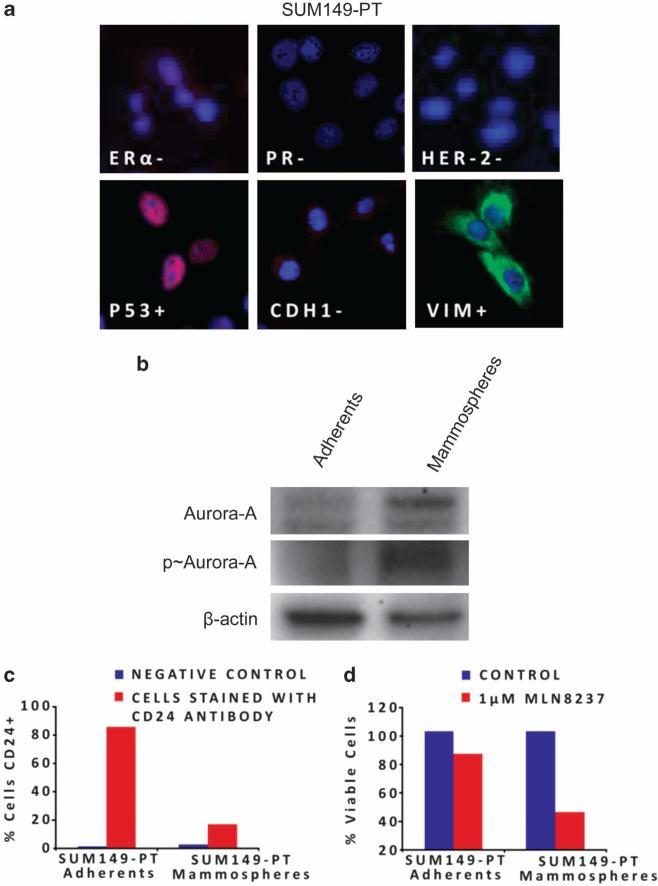
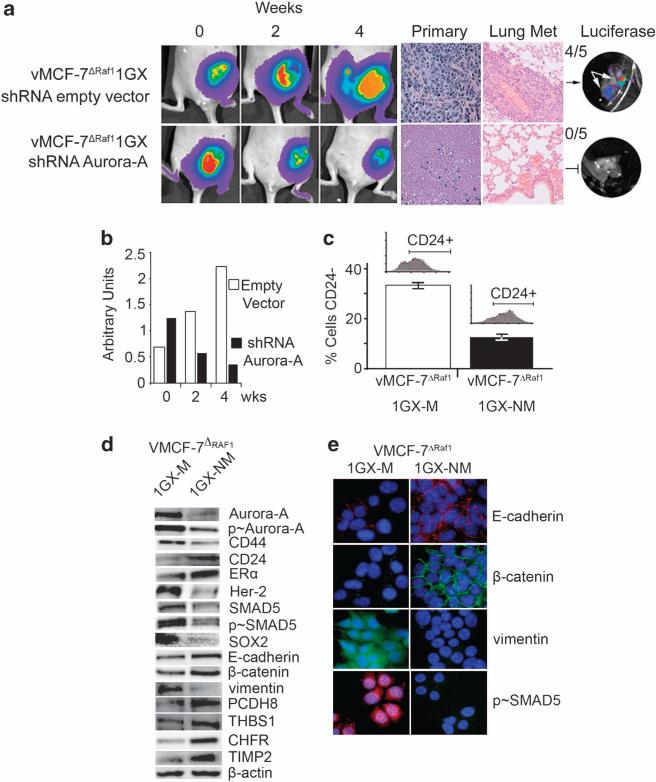
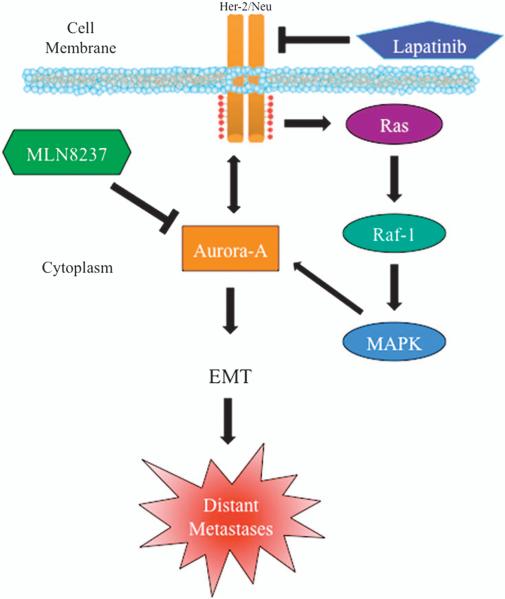
In this study, we demonstrate that constitutive activation of Raf-1 oncogenic signaling induces stabilization and accumulation of Aurora-A mitotic kinase that ultimately drives the transition from an epithelial to a highly invasive mesenchymal phenotype in estrogen receptor α-positive (ERα(+)) breast cancer cells. The transition from an epithelial- to a mesenchymal-like phenotype was characterized by reduced expression of ERα, HER-2/Neu overexpression and loss of CD24 surface receptor (CD24(-/low)). Importantly, expression of key epithelial-to-mesenchymal transition (EMT) markers and upregulation of the stemness gene SOX2 was linked to acquisition of stem cell-like properties such as the ability to form mammospheres in vitro and tumor self-renewal in vivo. Moreover, aberrant Aurora-A kinase activity induced phosphorylation and nuclear translocation of SMAD5, indicating a novel interplay between Aurora-A and SMAD5 signaling pathways in the development of EMT, stemness and ultimately tumor progression. Importantly, pharmacological and molecular inhibition of Aurora-A kinase activity restored a CD24(+) epithelial phenotype that was coupled to ERα expression, downregulation of HER-2/Neu, inhibition of EMT and impaired self-renewal ability, resulting in the suppression of distant metastases. Taken together, our findings show for the first time the causal role of Aurora-A kinase in the activation of EMT pathway responsible for the development of distant metastases in ERα(+) breast cancer cells. Moreover, this study has important translational implications because it highlights the mitotic kinase Aurora-A as a novel promising therapeutic target to selectively eliminate highly invasive cancer cells and improve the disease-free and overall survival of ERα(+) breast cancer patients resistant to conventional endocrine therapy.
Figures

References
-
- Basaran G, Devrim C, Caglar HB, Gulluoglu B, Kaya H, Seber S, et al. Clinical outcome of breast cancer patients with N3a (>/ = 10 positive lymph nodes) disease: has it changed over years? Med Oncol. 2010;28:726–732. - PubMed
-
- Chang H, Rha SY, Jeung HC, Im CK, Ahn JB, Kwon WS, et al. Association of the ABCB1 gene polymorphisms 2677G>T/A and 3435C>T with clinical outcomes of paclitaxel monotherapy in metastatic breast cancer patients. Ann Oncol. 2009;20:272–277. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
