

Genotype-phenotype correlation in 153 adult patients with congenital adrenal hyperplasia due to 21-hydroxylase deficiency: analysis of the United Kingdom Congenital adrenal Hyperplasia Adult Study Executive (CaHASE) cohort
- PMID: 23337727
- PMCID: PMC3651585
- DOI: 10.1210/jc.2012-3343
Genotype-phenotype correlation in 153 adult patients with congenital adrenal hyperplasia due to 21-hydroxylase deficiency: analysis of the United Kingdom Congenital adrenal Hyperplasia Adult Study Executive (CaHASE) cohort
Abstract
Context: In congenital adrenal hyperplasia (CAH) due to 21-hydroxylase deficiency, a strong genotype-phenotype correlation exists in childhood. However, similar data in adults are lacking.
Objective: The objective of the study was to test whether the severity of disease-causing CYP21A2 mutations influences the treatment and health status in adults with CAH.
Research design and methods: We analyzed the genotype in correlation with treatment and health status in 153 adults with CAH from the United Kingdom Congenital adrenal Hyperplasia Adult Study Executive cohort.
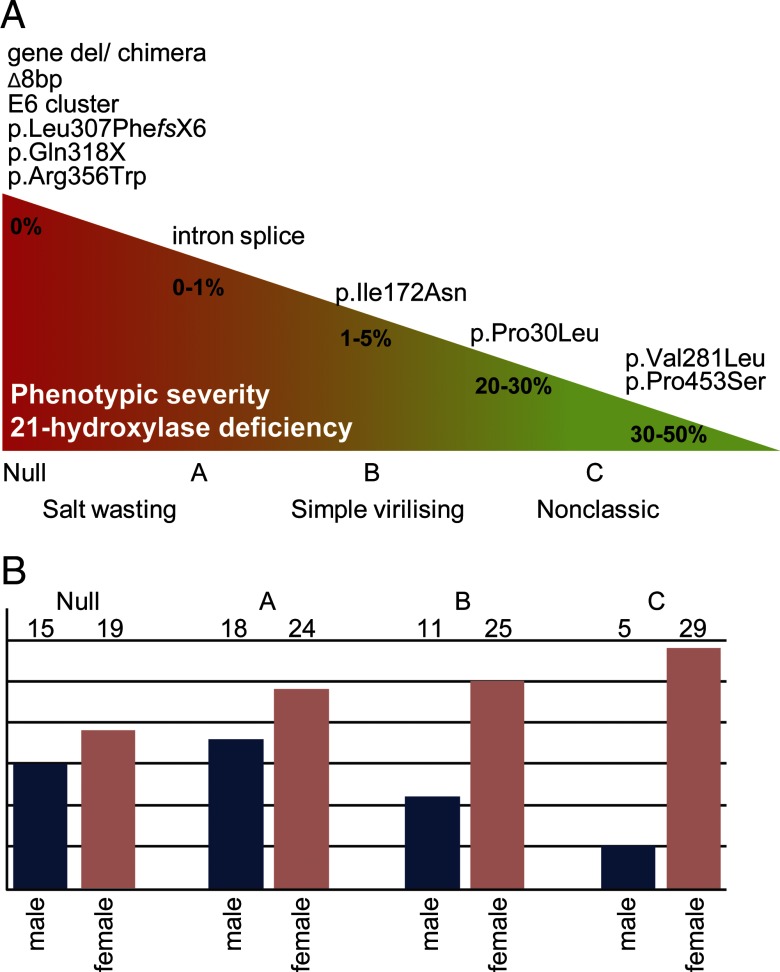
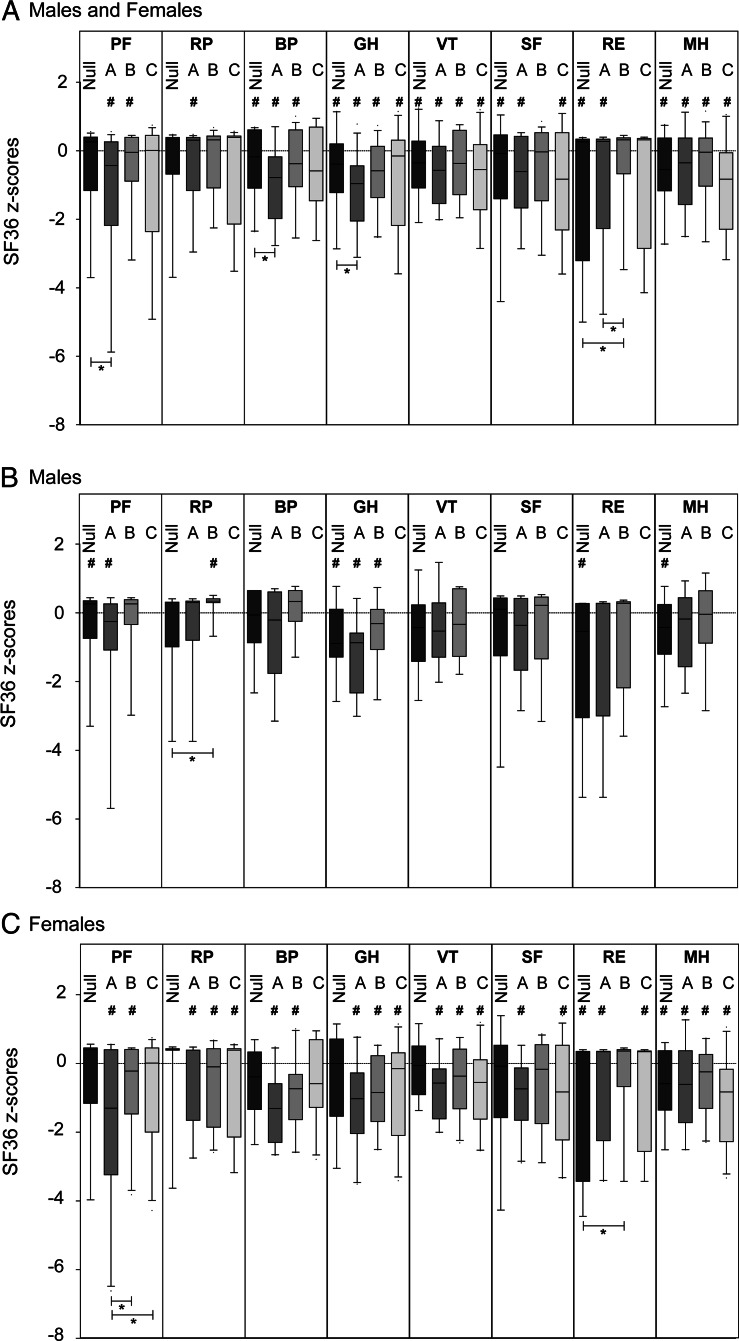
Results: CYP21A2 mutations were distributed similarly to previously reported case series. In 7 patients a mutation was identified on only 1 allele. Novel mutations were detected on 1.7% of alleles (5 of 306). Rare mutations were found on 2.3% of alleles (7 of 306). For further analysis, patients were categorized into CYP21A2 mutation groups according to predicted residual enzyme function: null (n = 34), A (n = 42), B (n = 36), C (n = 34), and D (n = 7). Daily glucocorticoid dose was highest in group null and lowest in group C. Fludrocortisone was used more frequently in patients with more severe genotypes. Except for lower female height in group B, no statistically significant associations between genotype and clinical parameters were found. Androgens, blood pressure, lipids, blood glucose, and homeostasis model assessment of insulin resistance were not different between groups. Subjective health status was similarly impaired across groups.
Conclusions: In adults with classic CAH and women with nonclassic CAH, there was a weak association between genotype and treatment, but health outcomes were not associated with genotype. The underrepresentation of males with nonclassic CAH may reflect that milder genotypes result in a milder condition that is neither diagnosed nor followed up in adulthood. Overall, our results suggest that the impaired health status of adults with CAH coming to medical attention is acquired rather than genetically determined and therefore could potentially be improved through modification of treatment.
Trial registration: ClinicalTrials.gov NCT00749593.
Figures
Comment in
-
Adrenal function: Adult CAH--does genotype correlate with phenotype?Nat Rev Endocrinol. 2013 Apr;9(4):187. doi: 10.1038/nrendo.2013.32. Epub 2013 Feb 12. Nat Rev Endocrinol. 2013. PMID: 23400016 No abstract available.
References
-
- White PC, Speiser PW. Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Endocr Rev. 2000;21:245–291 - PubMed
-
- Krone N, Dhir V, Ivison HE, Arlt W. Congenital adrenal hyperplasia and P450 oxidoreductase deficiency. Clin Endocrinol (Oxf). 2007;66:162–172 - PubMed
-
- Koppens PF, Hoogenboezem T, Degenhart HJ. Carriership of a defective tenascin-X gene in steroid 21-hydroxylase deficiency patients: TNXB-TNXA hybrids in apparent large-scale gene conversions. Hum Mol Genet. 2002;11:2581–2590 - PubMed
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
