

Safety and efficacy of velaglucerase alfa in Gaucher disease type 1 patients previously treated with imiglucerase
- PMID: 23339116
- PMCID: PMC3586535
- DOI: 10.1002/ajh.23383
Safety and efficacy of velaglucerase alfa in Gaucher disease type 1 patients previously treated with imiglucerase
Abstract
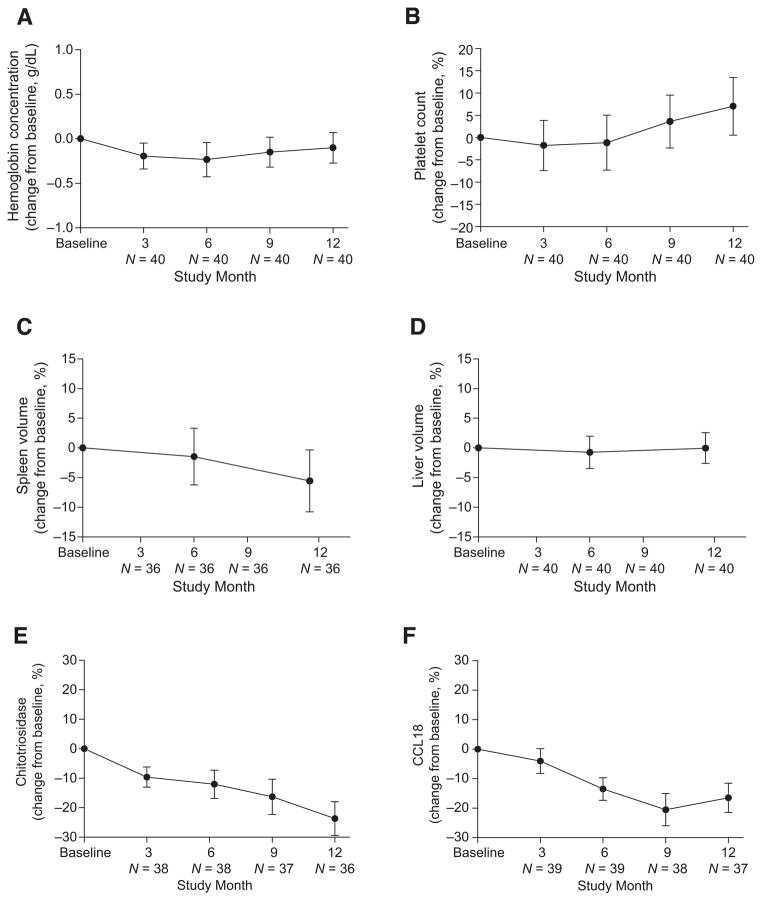
Velaglucerase alfa is a glucocerebrosidase produced by gene activation technology in a human fibroblast cell line (HT-1080), and it is indicated as an enzyme replacement therapy (ERT) for the treatment of Gaucher disease type 1 (GD1). This multicenter, open-label, 12-month study examined the safety and efficacy of velaglucerase alfa in patients with GD1 previously receiving imiglucerase. Eligible patients, ≥2 years old and clinically stable on imiglucerase therapy, were switched to velaglucerase alfa at a dose equal to their prior imiglucerase dose. Infusion durations were 1 hr every other week. Forty patients received velaglucerase alfa (18 male, 22 female; four previously splenectomized; age range 9-71 years). Velaglucerase alfa was generally well tolerated with most adverse events (AEs) of mild or moderate severity. The three most frequently reported AEs were headache (12 of 40 patients), arthralgia (9 of 40 patients), and nasopharyngitis (8 of 40 patients). No patients developed antibodies to velaglucerase alfa. There was one serious AE considered treatment-related: a grade 2 anaphylactoid reaction within 30 min of the first infusion. The patient withdrew; this was the only AE-related withdrawal. Hemoglobin concentrations, platelet counts, and spleen and liver volumes remained stable through 12 months. In conclusion, adult and pediatric patients with GD1, previously treated with imiglucerase, successfully transitioned to velaglucerase alfa, which was generally well tolerated and demonstrated efficacy over 12 months' treatment consistent with that observed in the velaglucerase alfa phase 3 clinical trial program.
Copyright © 2012 Wiley Periodicals, Inc.
Conflict of interest statement
The authors declare the following potential competing interests: Dr Zimran receives consulting fees from Protalix Biotherapeutics; has options in Protalix Biotherapeutics and sits on its Scientific Advisory Board; receives support from Genzyme for participation in the International Collaborative Gaucher Group (ICGG) Gaucher Registry; and receives honoraria from Shire HGT, Actelion, and Pfizer. He served as consultant to Shire HGT during the course of the trial (until October 2011). Dr Pastores is the recipient of research grants/support from Actelion, Amicus, Biomarin, Genzyme, Protalix, and Shire HGT, pharmaceutical/biotechnology companies engaged in drug development programs for the lysosomal storage disorders. Dr Hughes has received consulting fees, and travel and research grants and honoraria for speaking from Shire HGT, Genzyme, Protalix, and Amicus. Dr Mardach has no competing interests to declare. Dr Eng has received clinical trial research support and speaker support from Shire HGT. Dr Smith has received research grants from Genzyme, Shire HGT, and BioMarin, and consultant fees from Shire HGT and BioMarin. Dr Charrow has received consulting fees and honoraria from Genzyme, and consultant fees from Protalix/Pfizer. Dr Elstein received consulting fees and honoraria from Shire HGT. Dr Harmatz has provided consulting support to Shire HGT; received speaker’s honorarium and travel support from Shire HGT; participates in advisory boards (HOS) for Shire; provides consulting support to Biomarin; participated on advisory boards and has received research support and travel and speaker’s honorariums from Biomarin; and has received speaker’s honorarium from Genzyme. Dr Fernhoff received educational grants and research support from Shire HGT. Dr Rhead has received clinical trial support from Transkaryotic Therapies, Genzyme, Shire and Hyperion, and speaker fees from Ucyclyd. Dr Longo has received grant support from Shire HGT, Genzyme, Protalix/Pfizer, BioMarin, Amicus and Hyperion and consulting fees from BioMarin and Shire HGT. Dr Giraldo has received consulting fees from Shire HGT, Genzyme, and Actelion. Mr Zahrieh and Dr Crombez are employees of Shire HGT. Dr Grabowski has received consulting fees from Shire HGT, Genzyme, Pfizer, and Amicus Therapeutics. The Division of Human Genetics at Cincinnati Children’s Hospital Medical Center, for which Dr Grabowski serves as the Director, receives grants-in-aid for the conduct of basic and clinical research from Shire HGT and Genzyme, including the current study, as well as preclinical biopharmaceutical and small molecule studies that are unrelated to the studies presented here. Dr Grabowski does not hold stock or have stock options in any biopharmaceutical commercial concern.
Figures
References
-
- Grabowski GA, Petsko GA, Kolodny EH. The Online Metabolic and Molecular Bases of Inherited Diseases. [Accessed 8 May 2012];Chapter 146: Gaucher Disease. Available at: http://dx.doi.org/10.1036/ommbid.176. - DOI
-
- Tsuji S, Choudary PV, Martin BM, et al. Nucleotide sequence of cDNA containing the complete coding sequence for human lysosomal glucocerebrosidase. J Biol Chem. 1986;261:50–53. - PubMed
-
- Zimran A, Loveday K, Fratazzi C, et al. A pharmacokinetic analysis of a novel enzyme replacement therapy with Gene-Activated human glucocerebrosidase (GA-GCB) in patients with type 1 Gaucher disease. Blood Cells Mol Dis. 2007;39:115–118. - PubMed
-
- Zimran A, Altarescu G, Philips M, et al. Phase 1/2 and extension study of velaglucerase alfa replacement therapy in adults with type 1 Gaucher disease: 48-month experience. Blood. 2010;115:4651–4656. - PubMed
-
- Séllos-Moura M, Barzegar S, Pan L, et al. Development of a panel of highly sensitive, equivalent assays for detection of antibody responses to velaglucerase alfa or imiglucerase enzyme replacement therapy in patients with Gaucher disease. J Immunol Methods. 2011;373:45–53. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical