

Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone φ, ψ and side-chain χ(1) and χ(2) dihedral angles
- PMID: 23341755
- PMCID: PMC3549273
- DOI: 10.1021/ct300400x
Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone φ, ψ and side-chain χ(1) and χ(2) dihedral angles
Abstract
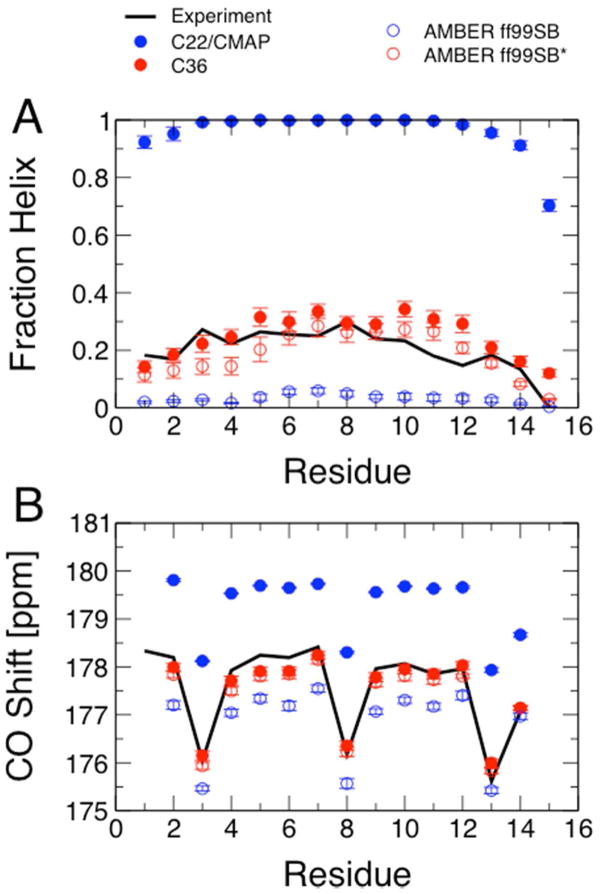
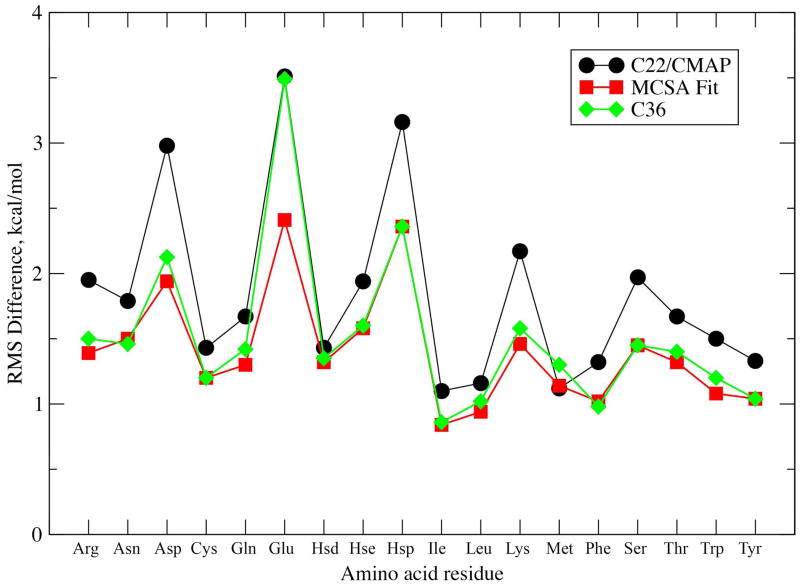
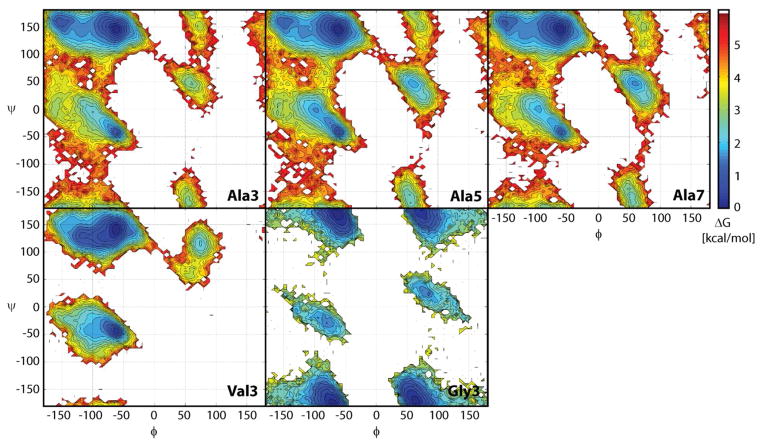
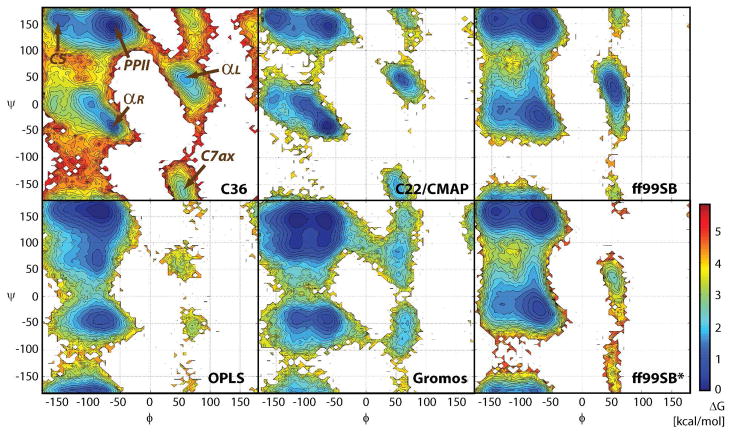
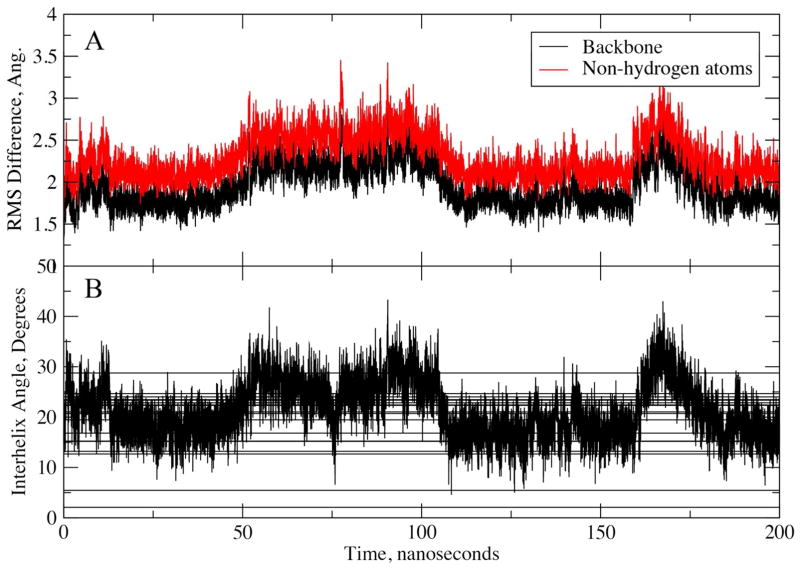
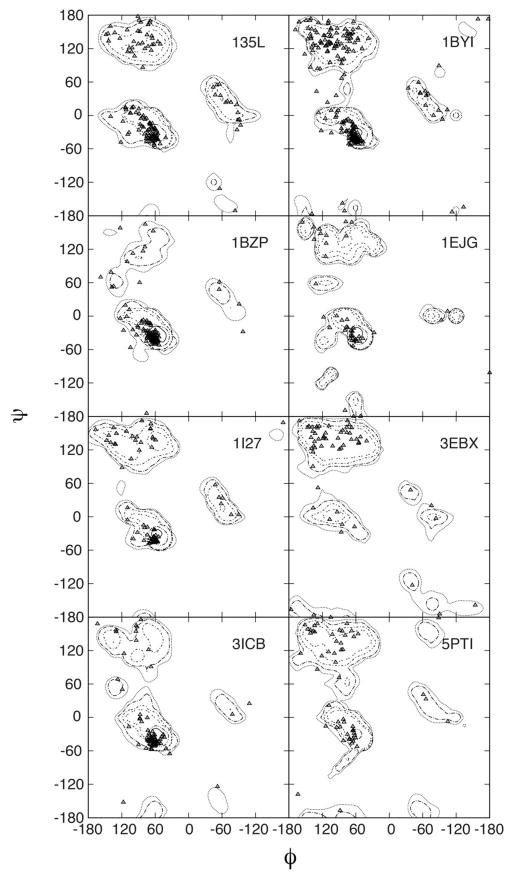
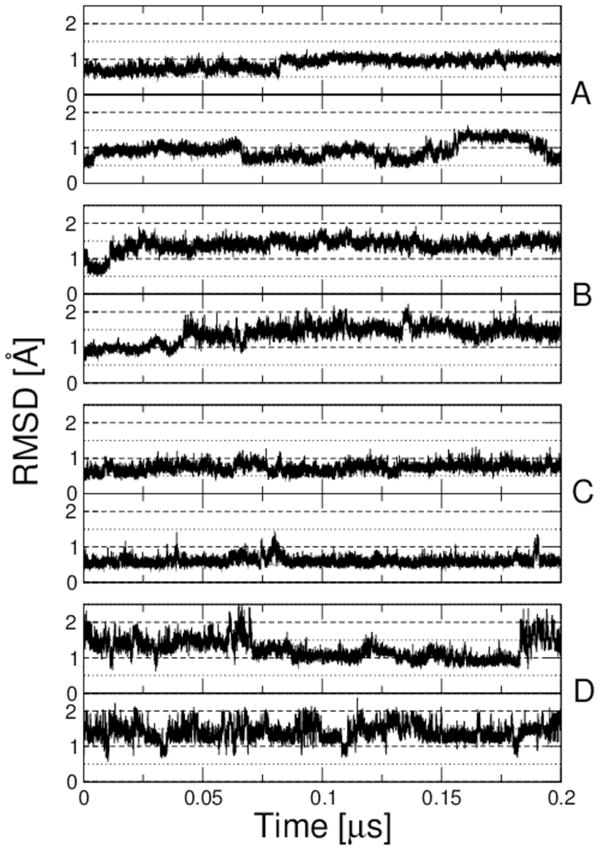
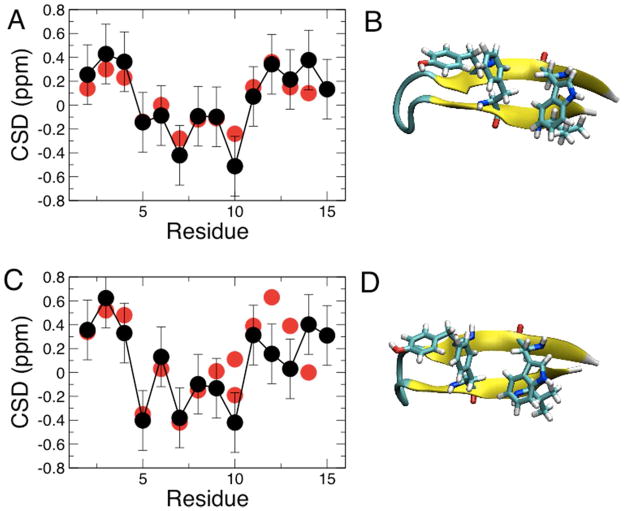
While the quality of the current CHARMM22/CMAP additive force field for proteins has been demonstrated in a large number of applications, limitations in the model with respect to the equilibrium between the sampling of helical and extended conformations in folding simulations have been noted. To overcome this, as well as make other improvements in the model, we present a combination of refinements that should result in enhanced accuracy in simulations of proteins. The common (non Gly, Pro) backbone CMAP potential has been refined against experimental solution NMR data for weakly structured peptides, resulting in a rebalancing of the energies of the α-helix and extended regions of the Ramachandran map, correcting the α-helical bias of CHARMM22/CMAP. The Gly and Pro CMAPs have been refitted to more accurate quantum-mechanical energy surfaces. Side-chain torsion parameters have been optimized by fitting to backbone-dependent quantum-mechanical energy surfaces, followed by additional empirical optimization targeting NMR scalar couplings for unfolded proteins. A comprehensive validation of the revised force field was then performed against data not used to guide parametrization: (i) comparison of simulations of eight proteins in their crystal environments with crystal structures; (ii) comparison with backbone scalar couplings for weakly structured peptides; (iii) comparison with NMR residual dipolar couplings and scalar couplings for both backbone and side-chains in folded proteins; (iv) equilibrium folding of mini-proteins. The results indicate that the revised CHARMM 36 parameters represent an improved model for the modeling and simulation studies of proteins, including studies of protein folding, assembly and functionally relevant conformational changes.
Figures
References
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources