

Predicting mendelian disease-causing non-synonymous single nucleotide variants in exome sequencing studies
- PMID: 23341771
- PMCID: PMC3547823
- DOI: 10.1371/journal.pgen.1003143
Predicting mendelian disease-causing non-synonymous single nucleotide variants in exome sequencing studies
Abstract
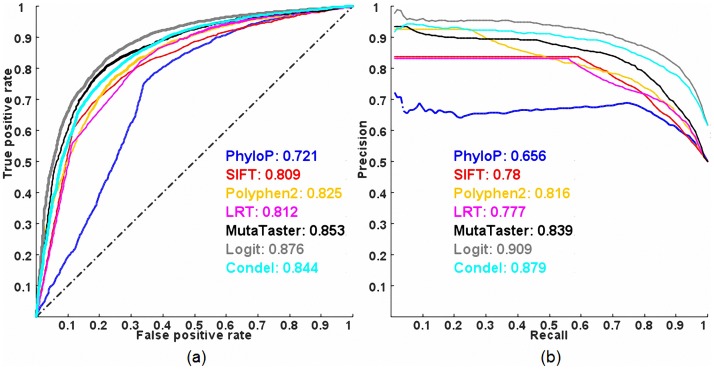
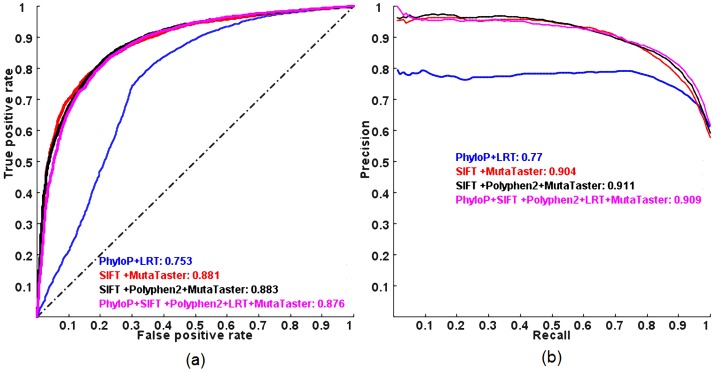
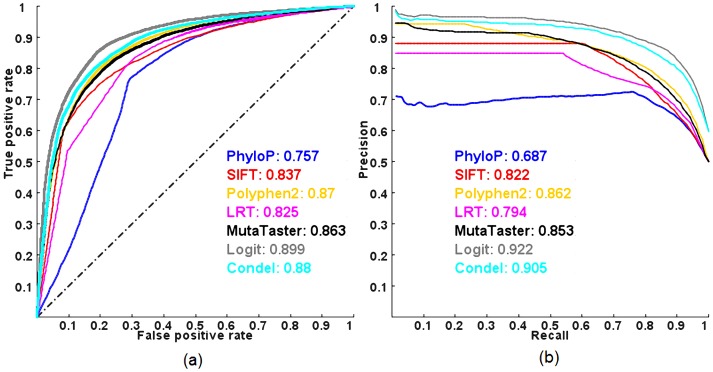
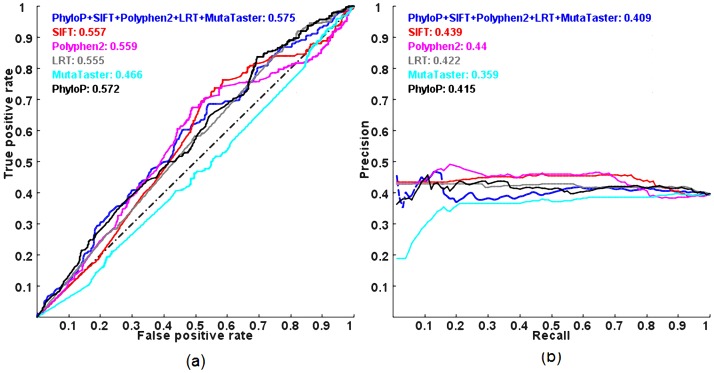
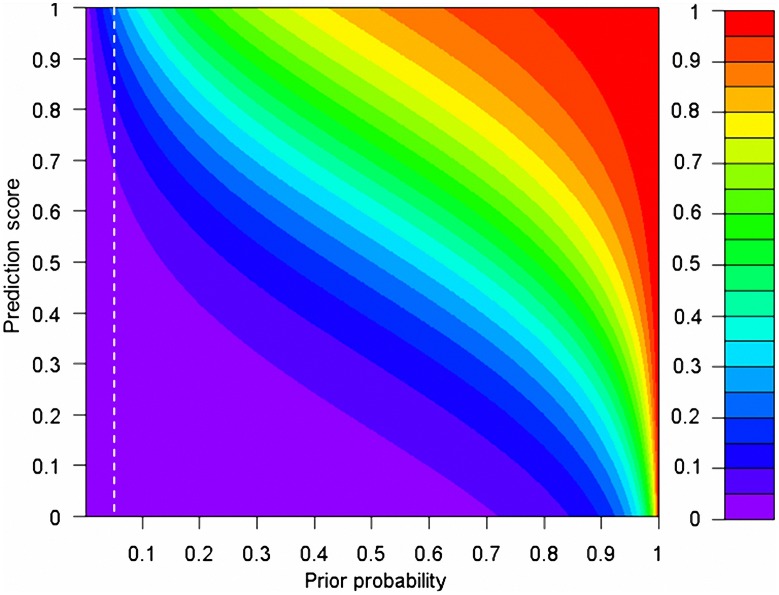
Exome sequencing is becoming a standard tool for mapping Mendelian disease-causing (or pathogenic) non-synonymous single nucleotide variants (nsSNVs). Minor allele frequency (MAF) filtering approach and functional prediction methods are commonly used to identify candidate pathogenic mutations in these studies. Combining multiple functional prediction methods may increase accuracy in prediction. Here, we propose to use a logit model to combine multiple prediction methods and compute an unbiased probability of a rare variant being pathogenic. Also, for the first time we assess the predictive power of seven prediction methods (including SIFT, PolyPhen2, CONDEL, and logit) in predicting pathogenic nsSNVs from other rare variants, which reflects the situation after MAF filtering is done in exome-sequencing studies. We found that a logit model combining all or some original prediction methods outperforms other methods examined, but is unable to discriminate between autosomal dominant and autosomal recessive disease mutations. Finally, based on the predictions of the logit model, we estimate that an individual has around 5% of rare nsSNVs that are pathogenic and carries ~22 pathogenic derived alleles at least, which if made homozygous by consanguineous marriages may lead to recessive diseases.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
