

Strategies for discovery and validation of methylated and hydroxymethylated DNA biomarkers
- PMID: 23342273
- PMCID: PMC3544446
- DOI: 10.1002/cam4.22
Strategies for discovery and validation of methylated and hydroxymethylated DNA biomarkers
Abstract
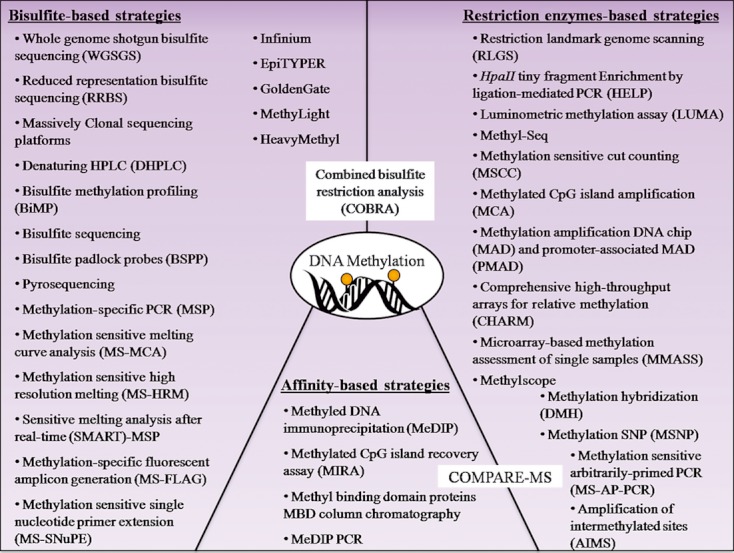
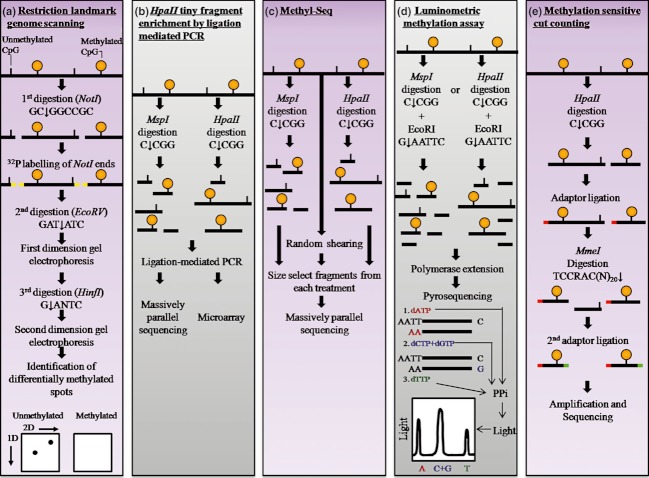
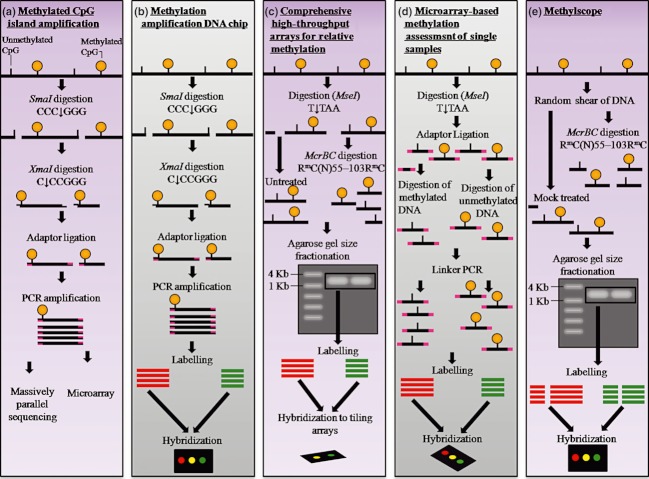
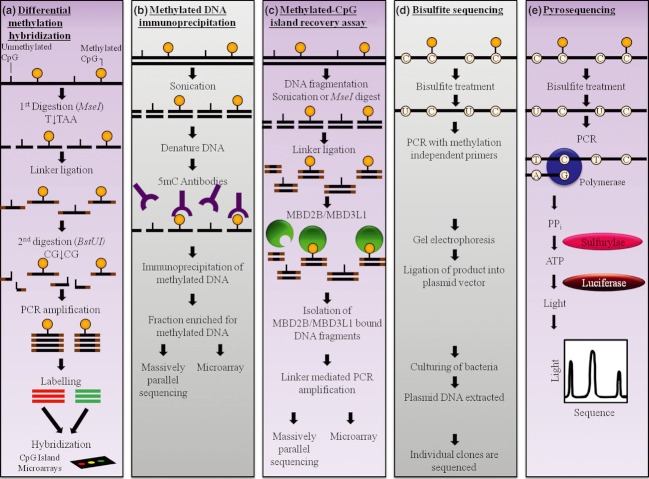
DNA methylation, consisting of the addition of a methyl group at the fifth-position of cytosine in a CpG dinucleotide, is one of the most well-studied epigenetic mechanisms in mammals with important functions in normal and disease biology. Disease-specific aberrant DNA methylation is a well-recognized hallmark of many complex diseases. Accordingly, various studies have focused on characterizing unique DNA methylation marks associated with distinct stages of disease development as they may serve as useful biomarkers for diagnosis, prognosis, prediction of response to therapy, or disease monitoring. Recently, novel CpG dinucleotide modifications with potential regulatory roles such as 5-hydroxymethylcytosine, 5-formylcytosine, and 5-carboxylcytosine have been described. These potential epigenetic marks cannot be distinguished from 5-methylcytosine by many current strategies and may potentially compromise assessment and interpretation of methylation data. A large number of strategies have been described for the discovery and validation of DNA methylation-based biomarkers, each with its own advantages and limitations. These strategies can be classified into three main categories: restriction enzyme digestion, affinity-based analysis, and bisulfite modification. In general, candidate biomarkers are discovered using large-scale, genome-wide, methylation sequencing, and/or microarray-based profiling strategies. Following discovery, biomarker performance is validated in large independent cohorts using highly targeted locus-specific assays. There are still many challenges to the effective implementation of DNA methylation-based biomarkers. Emerging innovative methylation and hydroxymethylation detection strategies are focused on addressing these gaps in the field of epigenetics. The development of DNA methylation- and hydroxymethylation-based biomarkers is an exciting and rapidly evolving area of research that holds promise for potential applications in diverse clinical settings.
Keywords: Affinity-based methylation analysis; bisulfite modification; hydroxymethylation; methylation-sensitive restriction enzymes; microarrays; next-generation sequencing.
Figures
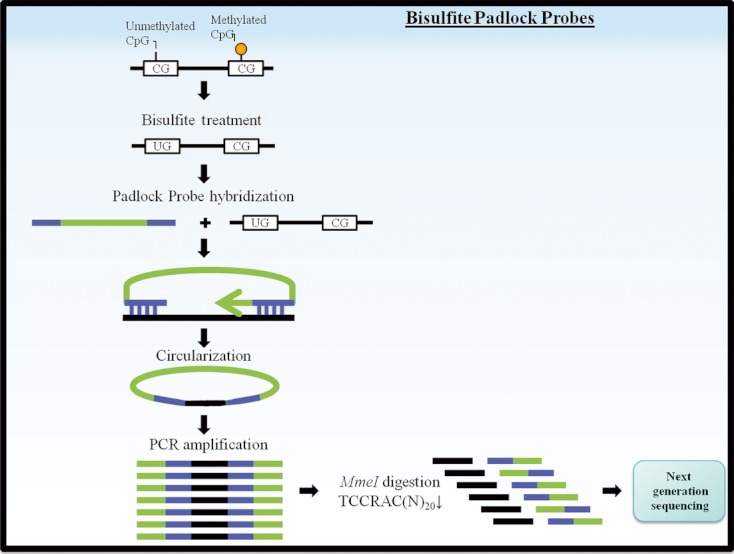
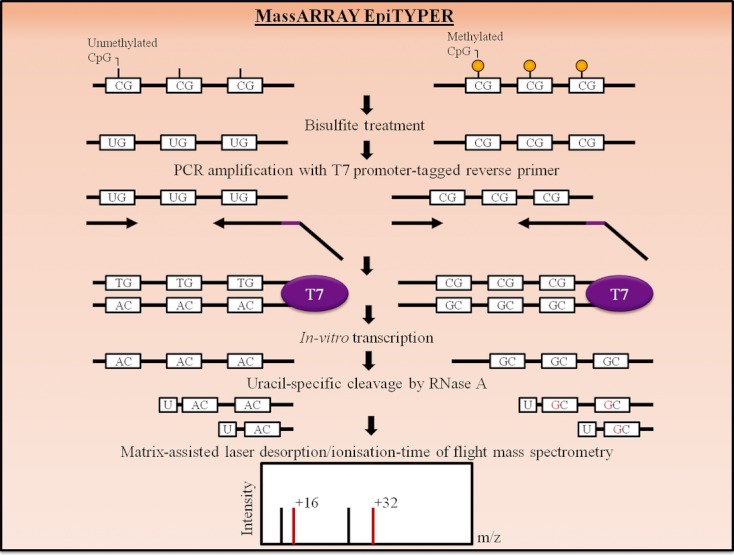
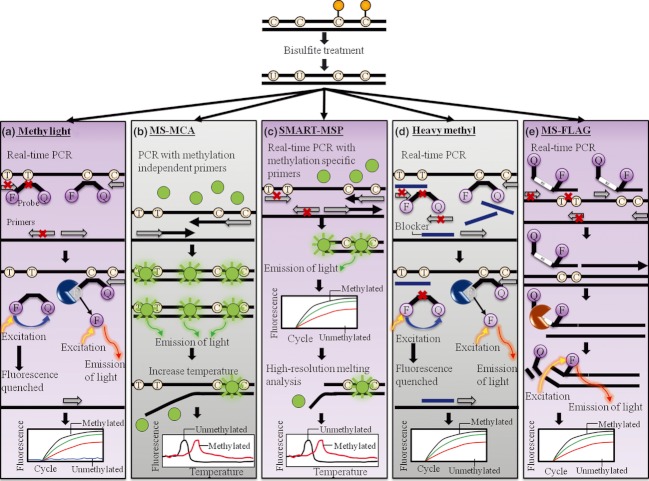
References
-
- Waddington CH. The epigenotype. Endeavour. 1942;1:18–20.
-
- Jablonka E, Lamb MJ. The changing concept of epigenetics. Ann. N. Y. Acad. Sci. 2002;981:82–96. - PubMed
-
- Bird A, Taggart M, Frommer M, Miller OJ, Macleod D. A fraction of the mouse genome that is derived from islands of nonmethylated, CpG-rich DNA. Cell. 1985;40:91–99. - PubMed
-
- Gardiner-Garden M, Frommer M. CpG islands in vertebrate genomes. J. Mol. Biol. 1987;196:261–282. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
