

Genome reannotation of the lizard Anolis carolinensis based on 14 adult and embryonic deep transcriptomes
- PMID: 23343042
- PMCID: PMC3561122
- DOI: 10.1186/1471-2164-14-49
Genome reannotation of the lizard Anolis carolinensis based on 14 adult and embryonic deep transcriptomes
Abstract
Background: The green anole lizard, Anolis carolinensis, is a key species for both laboratory and field-based studies of evolutionary genetics, development, neurobiology, physiology, behavior, and ecology. As the first non-avian reptilian genome sequenced, A. carolinesis is also a prime reptilian model for comparison with other vertebrate genomes. The public databases of Ensembl and NCBI have provided a first generation gene annotation of the anole genome that relies primarily on sequence conservation with related species. A second generation annotation based on tissue-specific transcriptomes would provide a valuable resource for molecular studies.
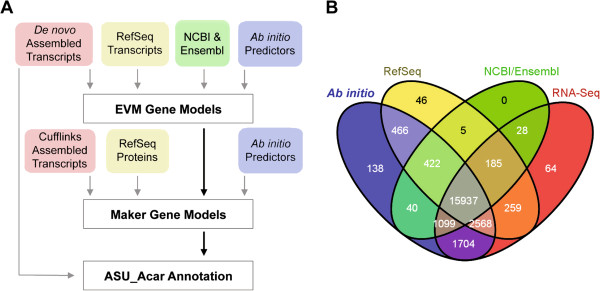
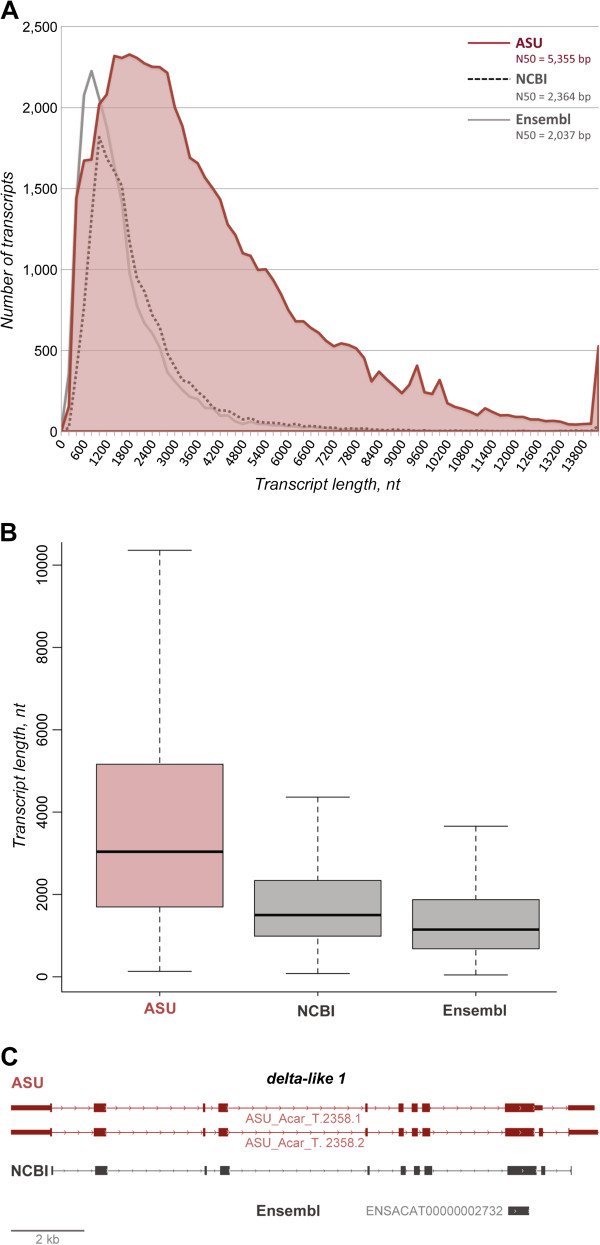
Results: Here we provide an annotation of the A. carolinensis genome based on de novo assembly of deep transcriptomes of 14 adult and embryonic tissues. This revised annotation describes 59,373 transcripts, compared to 16,533 and 18,939 currently for Ensembl and NCBI, and 22,962 predicted protein-coding genes. A key improvement in this revised annotation is coverage of untranslated region (UTR) sequences, with 79% and 59% of transcripts containing 5' and 3' UTRs, respectively. Gaps in genome sequence from the current A. carolinensis build (Anocar2.0) are highlighted by our identification of 16,542 unmapped transcripts, representing 6,695 orthologues, with less than 70% genomic coverage.
Conclusions: Incorporation of tissue-specific transcriptome sequence into the A. carolinensis genome annotation has markedly improved its utility for comparative and functional studies. Increased UTR coverage allows for more accurate predicted protein sequence and regulatory analysis. This revised annotation also provides an atlas of gene expression specific to adult and embryonic tissues.
Figures
Similar articles
-
Reptilian Transcriptomes v2.0: An Extensive Resource for Sauropsida Genomics and Transcriptomics.Genome Biol Evol. 2015 Jul 1;7(6):1827-41. doi: 10.1093/gbe/evv106. Genome Biol Evol. 2015. PMID: 26133641 Free PMC article.
-
A high-quality annotated transcriptome of swine peripheral blood.BMC Genomics. 2017 Jun 24;18(1):479. doi: 10.1186/s12864-017-3863-7. BMC Genomics. 2017. PMID: 28646867 Free PMC article.
-
A garter snake transcriptome: pyrosequencing, de novo assembly, and sex-specific differences.BMC Genomics. 2010 Dec 7;11:694. doi: 10.1186/1471-2164-11-694. BMC Genomics. 2010. PMID: 21138572 Free PMC article.
-
Evolutionary dynamics of coding and non-coding transcriptomes.Nat Rev Genet. 2014 Nov;15(11):734-48. doi: 10.1038/nrg3802. Epub 2014 Oct 9. Nat Rev Genet. 2014. PMID: 25297727 Review.
-
Decoding the Equine Genome: Lessons from ENCODE.Genes (Basel). 2021 Oct 27;12(11):1707. doi: 10.3390/genes12111707. Genes (Basel). 2021. PMID: 34828313 Free PMC article. Review.
Cited by
-
The Genome 10K Project: a way forward.Annu Rev Anim Biosci. 2015;3:57-111. doi: 10.1146/annurev-animal-090414-014900. Annu Rev Anim Biosci. 2015. PMID: 25689317 Free PMC article. Review.
-
Chromosome-Level Assembly of the Common Lizard (Zootoca vivipara) Genome.Genome Biol Evol. 2020 Nov 3;12(11):1953-1960. doi: 10.1093/gbe/evaa161. Genome Biol Evol. 2020. PMID: 32835354 Free PMC article.
-
Differential expression of conserved and novel microRNAs during tail regeneration in the lizard Anolis carolinensis.BMC Genomics. 2016 May 5;17:339. doi: 10.1186/s12864-016-2640-3. BMC Genomics. 2016. PMID: 27150582 Free PMC article.
-
MAKER-P: a tool kit for the rapid creation, management, and quality control of plant genome annotations.Plant Physiol. 2014 Feb;164(2):513-24. doi: 10.1104/pp.113.230144. Epub 2013 Dec 4. Plant Physiol. 2014. PMID: 24306534 Free PMC article.
-
OGS2: genome re-annotation of the jewel wasp Nasonia vitripennis.BMC Genomics. 2016 Aug 25;17(1):678. doi: 10.1186/s12864-016-2886-9. BMC Genomics. 2016. PMID: 27561358 Free PMC article.
References
-
- Rhind N, Chen Z, Yassour M, Thompson DA, Haas BJ, Habib N, Wapinski I, Roy S, Lin MF, Heiman DI, Young SK, Furuya K, Guo Y, Pidoux A, Chen HM, Robbertse B, Goldberg JM, Aoki K, Bayne EH, Berlin AM, Desjardins CA, Dobbs E, Dukaj L, Fan L, FitzGerald MG, French C, Gujja S, Hansen K, Keifenheim D, Levin JZ, Mosher RA, Muller CA, Pfiffner J, Priest M, Russ C, Smialowska A, Swoboda P, Sykes SM, Vaughn M, Vengrova S, Yoder R, Zeng Q, Allshire R, Baulcombe D, Birren BW, Brown W, Ekwall K, Kellis M, Leatherwood J, Levin H, Margalit H, Martienssen R, Nieduszynski CA, Spatafora JW, Friedman N, Dalgaard JZ, Baumann P, Niki H, Regev A, Nusbaum C. Comparative Functional Genomics of the Fission Yeasts. Science. 2011;332:930–936. doi: 10.1126/science.1203357. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
