

Impact of geometry optimization on base-base stacking interaction energies in the canonical A- and B-forms of DNA
- PMID: 23343365
- PMCID: PMC3579007
- DOI: 10.1021/jp308364d
Impact of geometry optimization on base-base stacking interaction energies in the canonical A- and B-forms of DNA
Abstract
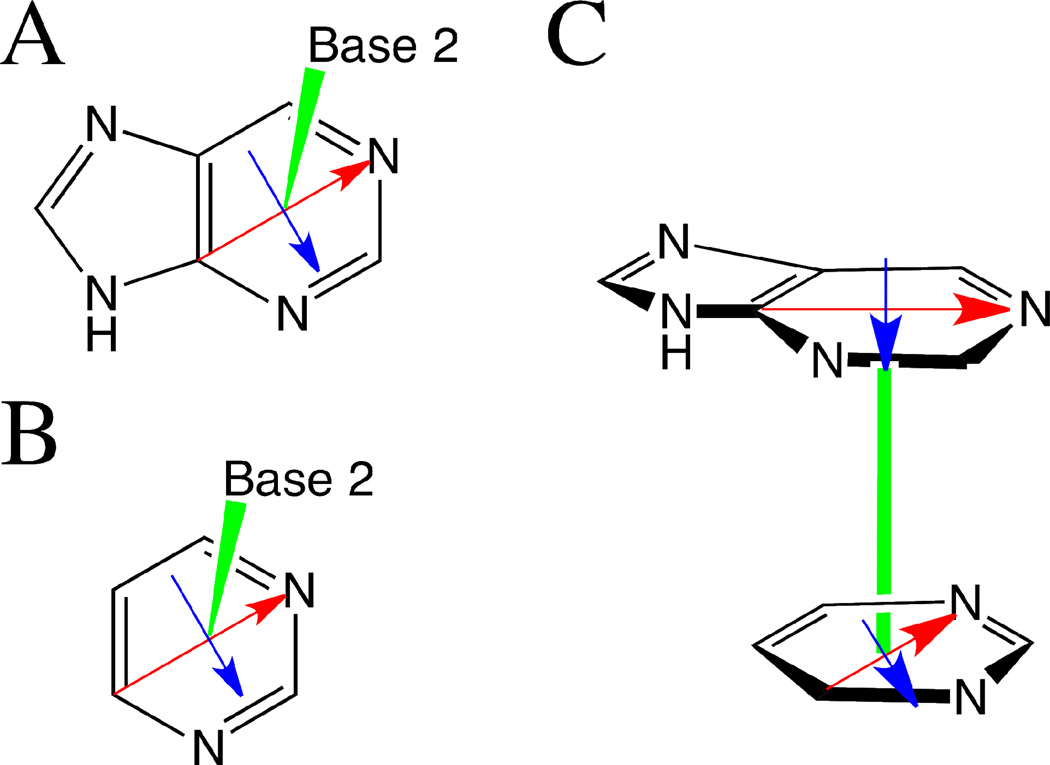
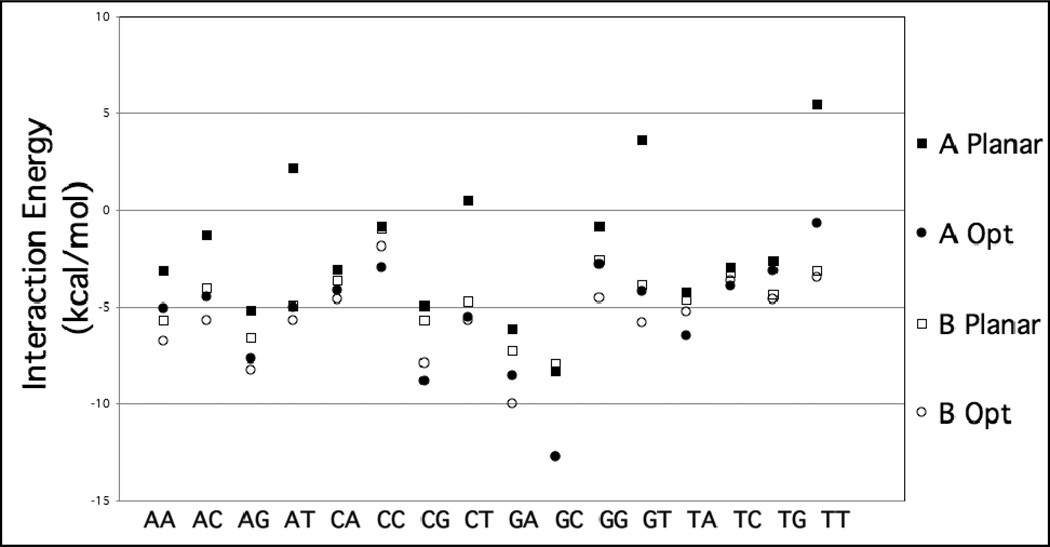
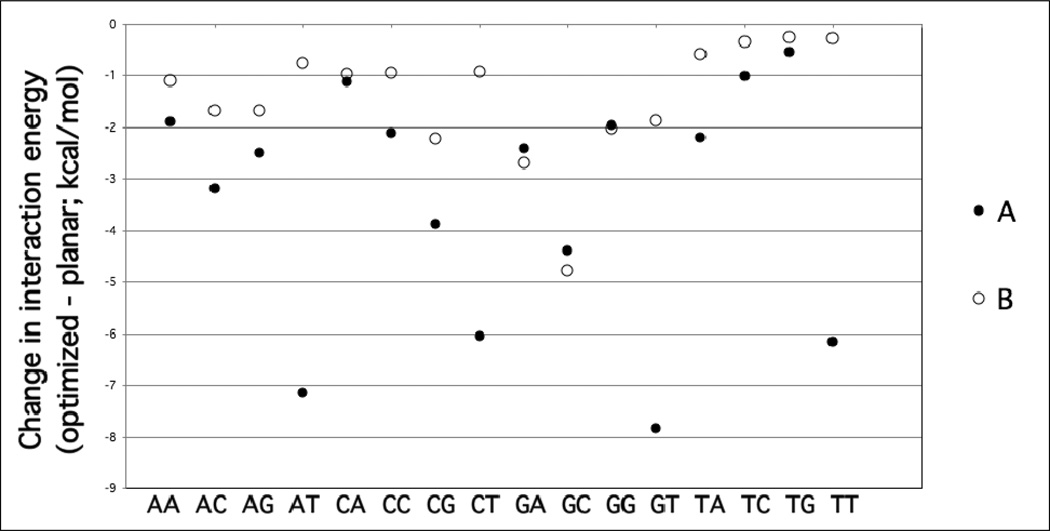
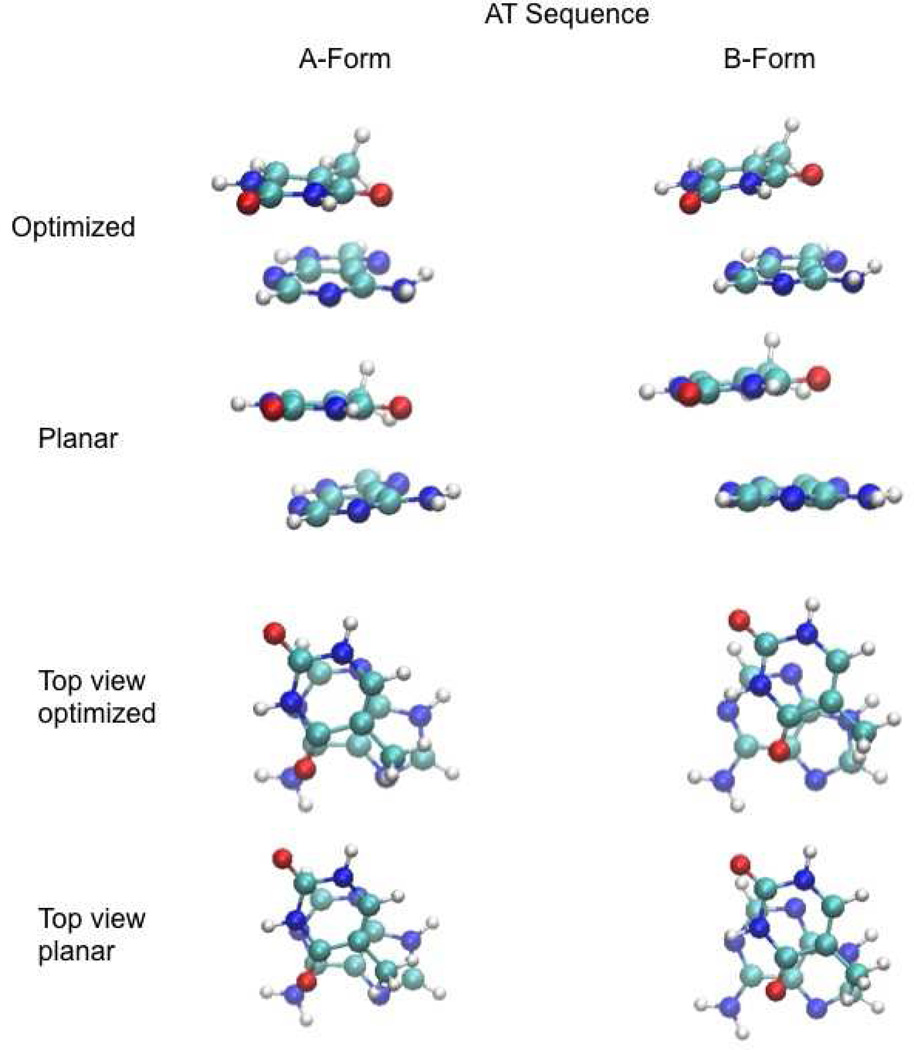
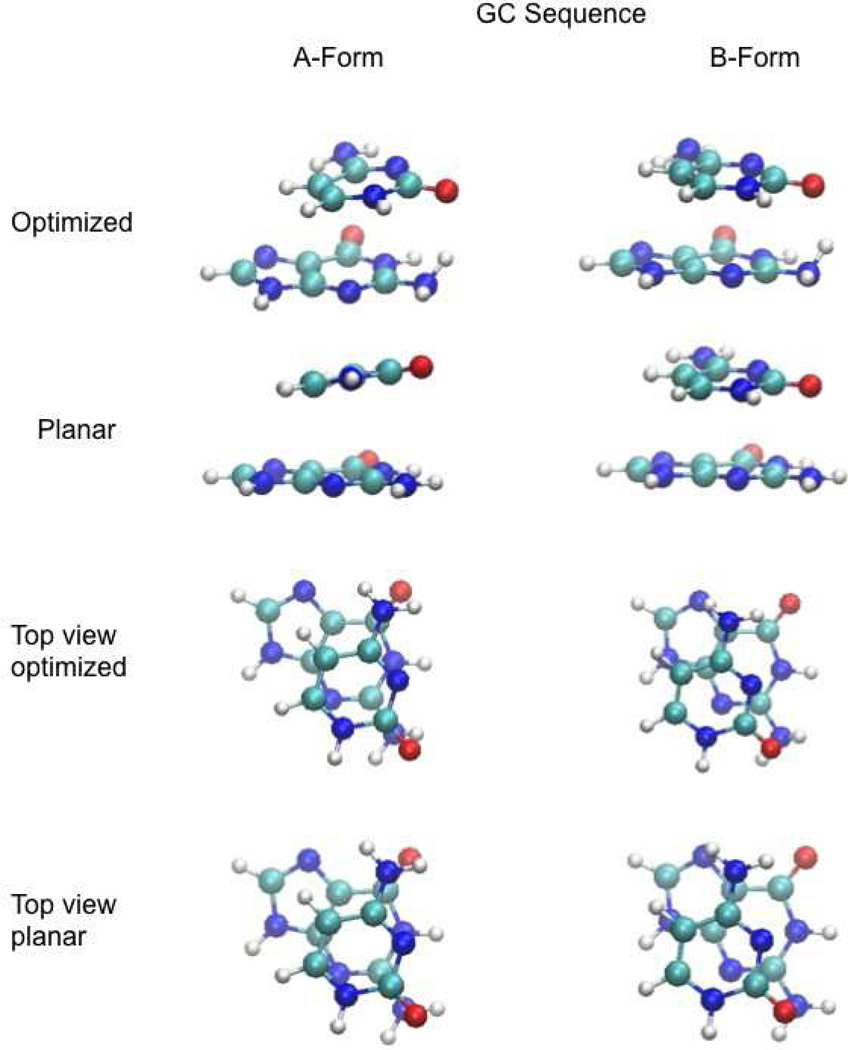
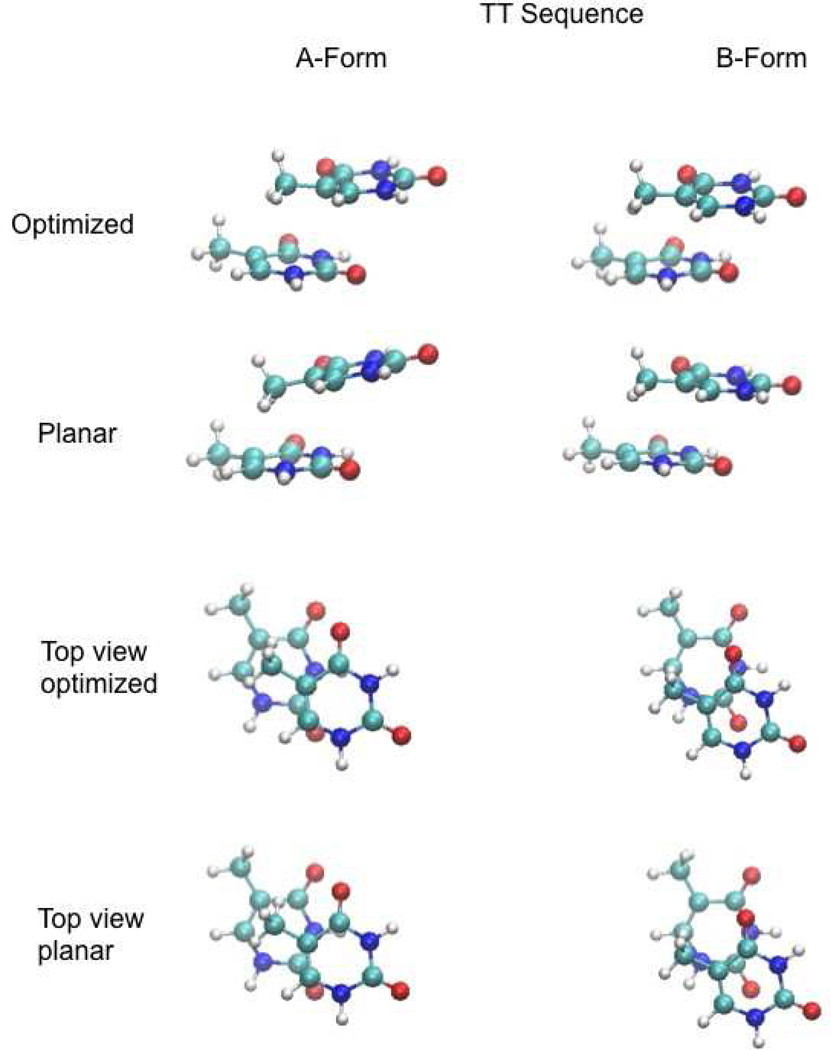
Base stacking is known to make an important contribution to the stability of DNA and RNA, and accordingly, significant efforts are ongoing to calculate stacking energies using ab initio quantum mechanical methods. To date, impressive improvements have been made in the model chemistries used to perform stacking energy calculations, including extensions that include robust treatments of electron correlation with extended basis sets, as required to treat interactions where dispersion makes a significant contribution. However, those efforts typically use rigid monomer geometries when calculating the interaction energies. To overcome this, in the present work, we describe a novel internal coordinate definition that allows the relative, intermolecular orientation of stacked base monomers to be constrained during geometry optimizations while allowing full optimization of the intramolecular degrees of freedom. Use of the novel reference frame to calculate the impact of full geometry optimization versus constraining the bases to be planar on base monomer stacking energies, combined with density-fitted, spin-component scaling MP2 treatment of electron correlation, shows that full optimization makes the average stacking energy more favorable by -3.4 and -1.5 kcal/mol for the canonical A and B conformations of the 16 5' to 3' base stacked monomers. Thus, treatment of geometry optimization impacts the stacking energies to an extent similar to or greater than the impact of current state of the art increases in the rigor of the model chemistry itself used to treat base stacking. Results also indicate that stacking favors the B-form of DNA, though the average difference versus the A-form decreases from -2.6 to -0.6 kcal/mol when the intramolecular geometry is allowed to fully relax. However, stacking involving cytosine is shown to favor the A-form of DNA, with that contribution generally larger in the fully optimized bases. The present results show the importance of allowing geometry optimization, as well as properly treating the appropriate model chemistry, in studies of nucleic acid base stacking.
Figures

References
-
- Olson WK, Zhurkin VB. Current Opinion in Structural Biology. 2000;10:286–297. - PubMed
-
- Ivanov VI, Krylov DY. Methods Enzymol. 1992;211:111–127. - PubMed
-
- Franklin RE, Gosling RG. Nature. 1953;171:740–741. - PubMed
-
- Pohl FM, Jovin TM. J. Mol. Biol. 1972;67:375–396. - PubMed
-
- Leslie AGW, Arnott S, Chandrasekaran R, Ratliff RL. J. Mol. Biol. 1980;143:49–72. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
