

Comment
doi: 10.1172/JCI66882.
Epub 2013 Jan 25.
Ubiquitylation and the pathogenesis of hypertension
Affiliations
- PMID: 23348731
- PMCID: PMC3561829
- DOI: 10.1172/JCI66882
Item in Clipboard
Comment
Ubiquitylation and the pathogenesis of hypertension
J Clin Invest.
2013 Feb.
Abstract
Liddle syndrome is monogenic hypertension caused by mutations in the epithelial Na+ channel (ENaC) that interfere with its ubiquitylation by Nedd4-2. In this issue, Ronzaud and colleagues found that deleting Nedd4-2 from kidney tubules in adult mice led to ENaC accumulation, but not at the plasma membrane, as predicted from current models. Instead, abundance of the sodium chloride transporter NCC increased at the plasma membrane, and the mice have some features of increased NCC activity. Together, the results suggest that defective ubiquitylation of ENaC by Nedd4-2 may not fully explain Liddle syndrome and that Nedd4-2 modulates NCC more strongly.
Figures
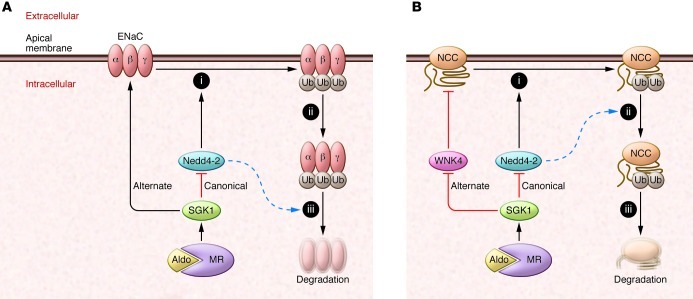
(A) Aldosterone (Aldo) interaction with the mineralocorticoid receptor (MR) induces SGK1 activity, which suppresses the ubiquitin ligase activity of Nedd4-2. Nedd4-2 normally catalyzes the addition of ubiquitin moieties (Ub) to ENaC (step i). Ubiquitylated ENaC is then removed from the plasma membrane (step ii) and undergoes degradation (or recycling) inside the cell (step iii). Poorly defined alternate pathways for SGK1 to stimulate ENaC independently of Nedd4-2 also exist. Ronzaud and colleagues (8) found that Nedd4-2 deletion increased in intracellular ENaC, suggesting that Nedd4-2 may affect step iii (dotted blue arrow) more than step ii. (B) Similar signaling pathway to NCC. An alternative pathway for SGK1 activation of NCC may involve WNK4, as described in text. In contrast with effects on ENaC, NCC abundance was increased at the plasma membrane by Nedd4-2 deletion, suggesting a defect at step ii, as shown by the dotted blue arrow. Note that sites of ubiquitylation on NCC have not been defined.
Comment on
-
Renal tubular NEDD4-2 deficiency causes NCC-mediated salt-dependent hypertension.J Clin Invest. 2013 Feb;123(2):657-65. doi: 10.1172/JCI61110. Epub 2013 Jan 25. J Clin Invest. 2013. PMID: 23348737 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
