

67-kDa laminin receptor increases cGMP to induce cancer-selective apoptosis
- PMID: 23348740
- PMCID: PMC3561824
- DOI: 10.1172/JCI64768
67-kDa laminin receptor increases cGMP to induce cancer-selective apoptosis
Abstract
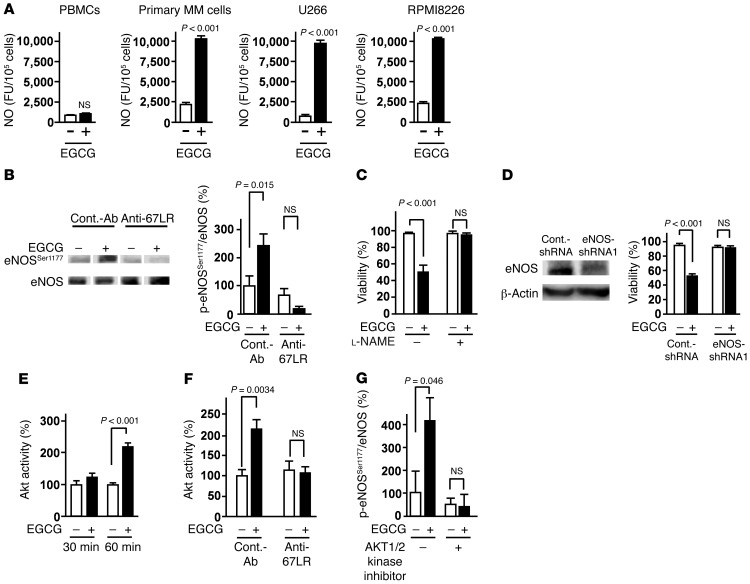
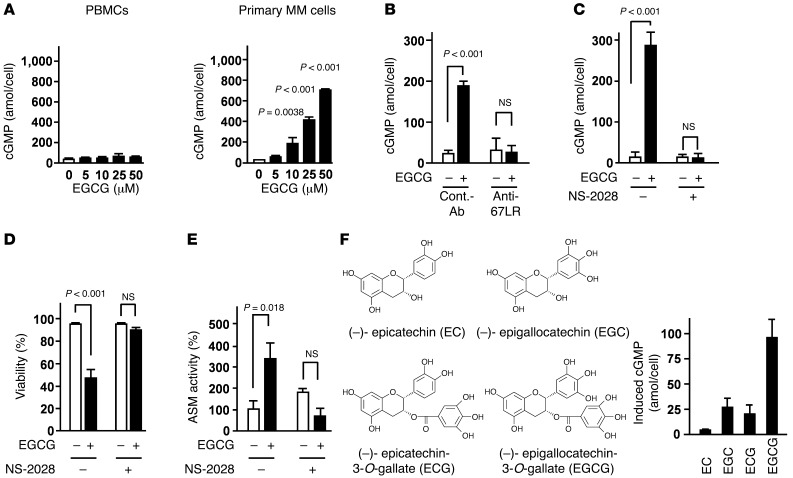
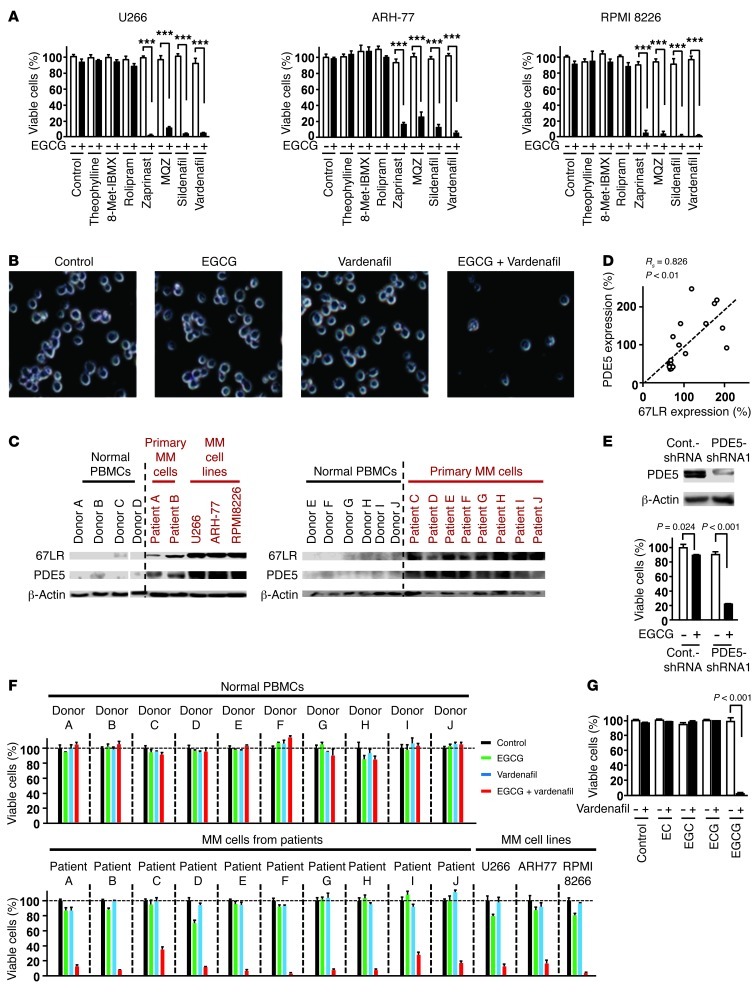
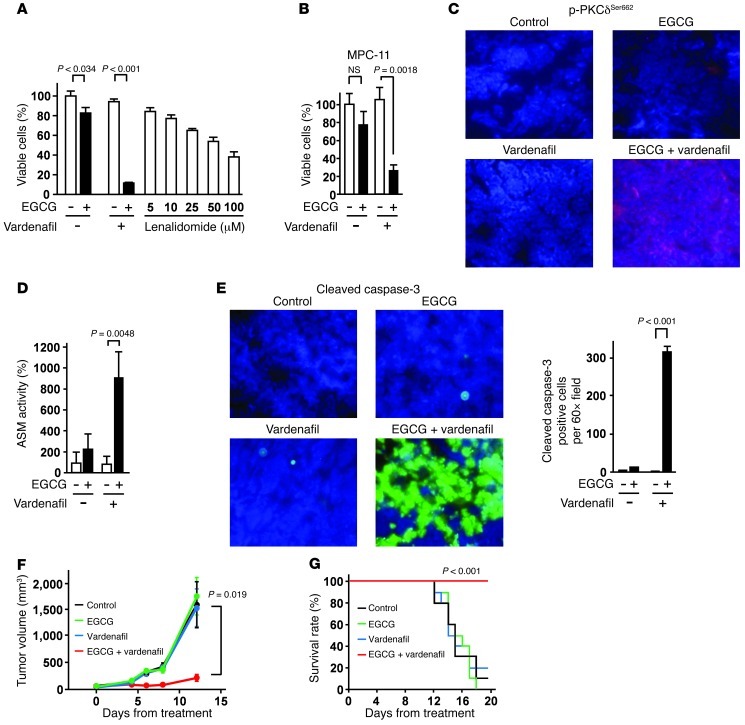
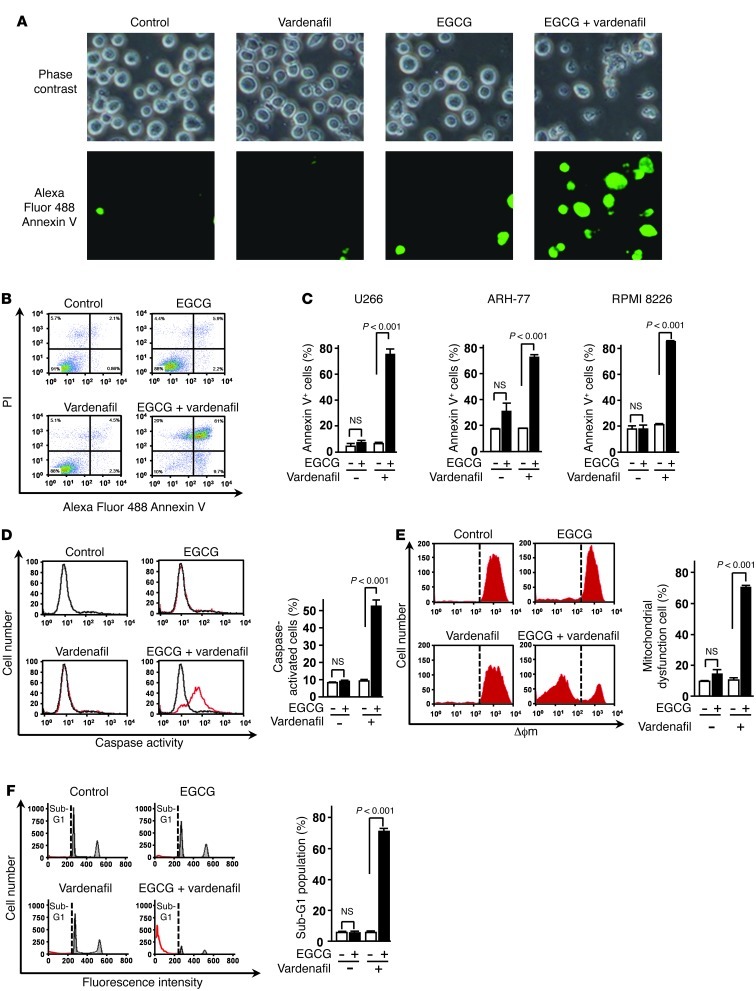
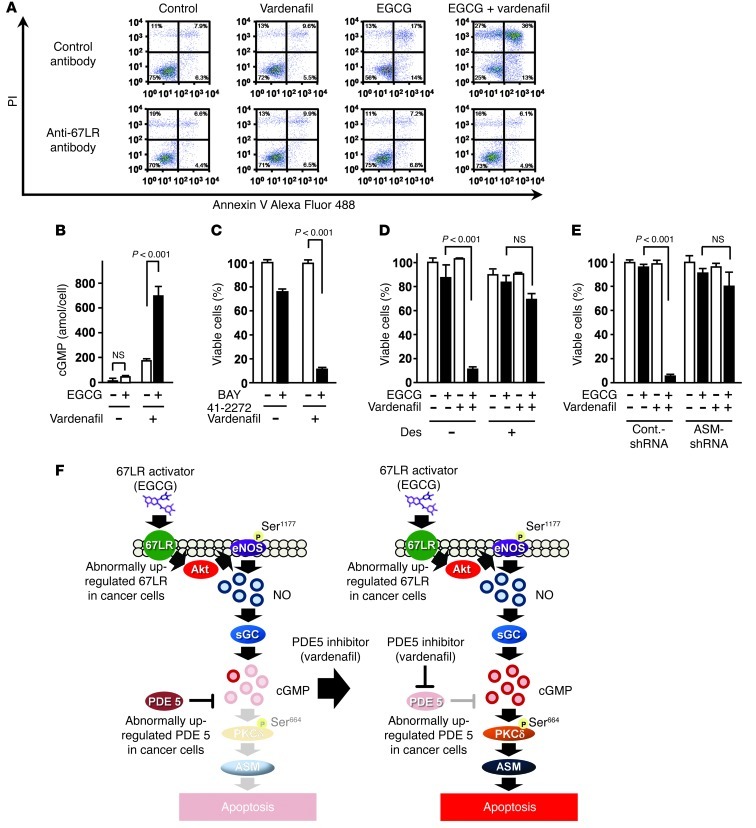
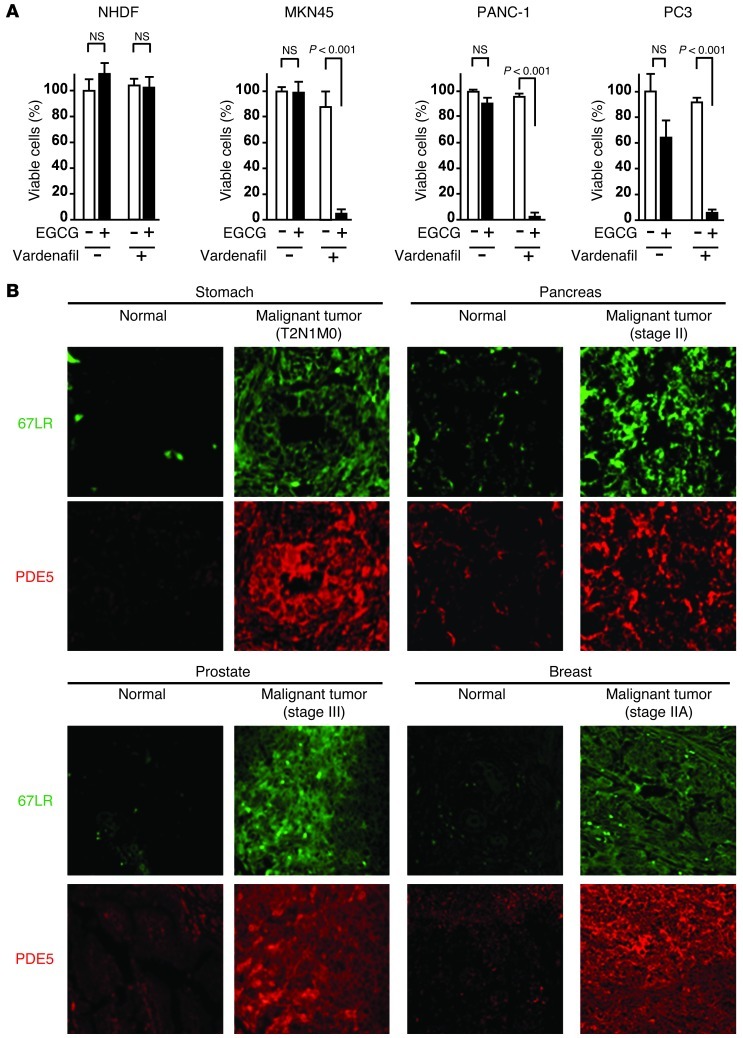
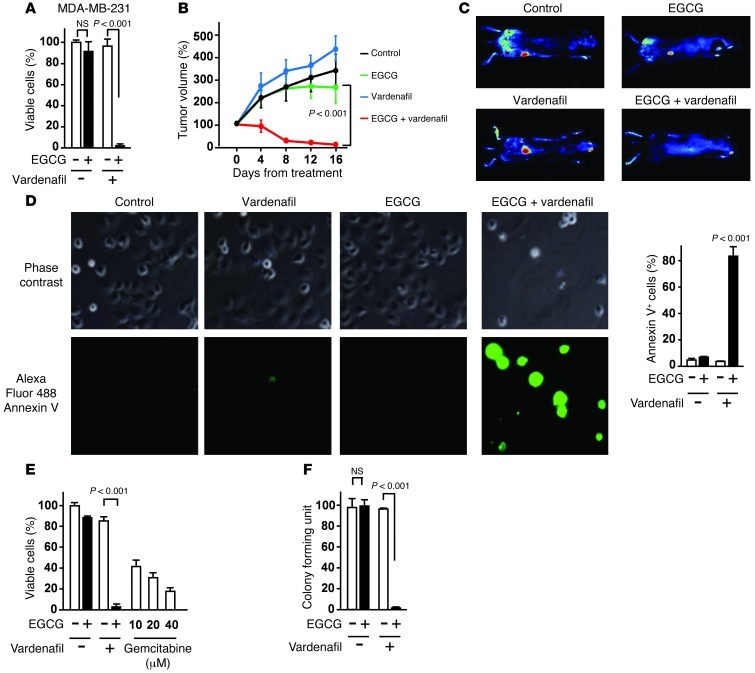
The 67-kDa laminin receptor (67LR) is a laminin-binding protein overexpressed in various types of cancer, including bile duct carcinoma, colorectal carcinoma, cervical cancer, and breast carcinoma. 67LR plays a vital role in growth and metastasis of tumor cells and resistance to chemotherapy. Here, we show that 67LR functions as a cancer-specific death receptor. In this cell death receptor pathway, cGMP initiated cancer-specific cell death by activating the PKCδ/acid sphingomyelinase (PKCδ/ASM) pathway. Furthermore, upregulation of cGMP was a rate-determining process of 67LR-dependent cell death induced by the green tea polyphenol (-)-epigallocatechin-3-O-gallate (EGCG), a natural ligand of 67LR. We found that phosphodiesterase 5 (PDE5), a negative regulator of cGMP, was abnormally expressed in multiple cancers and attenuated 67LR-mediated cell death. Vardenafil, a PDE5 inhibitor that is used to treat erectile dysfunction, significantly potentiated the EGCG-activated 67LR-dependent apoptosis without affecting normal cells and prolonged the survival time in a mouse xenograft model. These results suggest that PDE5 inhibitors could be used to elevate cGMP levels to induce 67LR-mediated, cancer-specific cell death.
Figures
Comment in
-
Cancer therapy combination: green tea and a phosphodiesterase 5 inhibitor?J Clin Invest. 2013 Feb;123(2):556-8. doi: 10.1172/JCI67589. Epub 2013 Jan 25. J Clin Invest. 2013. PMID: 23348734 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
