

Blockade of NOX2 and STIM1 signaling limits lipopolysaccharide-induced vascular inflammation
- PMID: 23348743
- PMCID: PMC3561818
- DOI: 10.1172/JCI65647
Blockade of NOX2 and STIM1 signaling limits lipopolysaccharide-induced vascular inflammation
Abstract
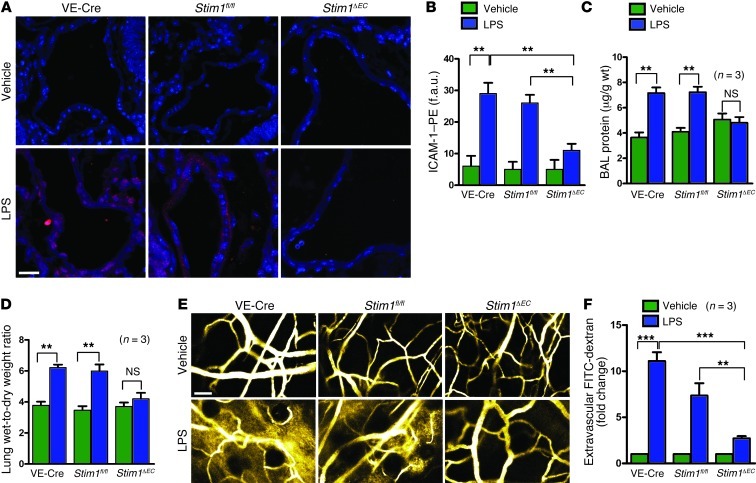
During sepsis, acute lung injury (ALI) results from activation of innate immune cells and endothelial cells by endotoxins, leading to systemic inflammation through proinflammatory cytokine overproduction, oxidative stress, and intracellular Ca2+ overload. Despite considerable investigation, the underlying molecular mechanism(s) leading to LPS-induced ALI remain elusive. To determine whether stromal interaction molecule 1-dependent (STIM1-dependent) signaling drives endothelial dysfunction in response to LPS, we investigated oxidative and STIM1 signaling of EC-specific Stim1-knockout mice. Here we report that LPS-mediated Ca2+ oscillations are ablated in ECs deficient in Nox2, Stim1, and type II inositol triphosphate receptor (Itpr2). LPS-induced nuclear factor of activated T cells (NFAT) nuclear accumulation was abrogated by either antioxidant supplementation or Ca2+ chelation. Moreover, ECs lacking either Nox2 or Stim1 failed to trigger store-operated Ca2+ entry (SOCe) and NFAT nuclear accumulation. LPS-induced vascular permeability changes were reduced in EC-specific Stim1-/- mice, despite elevation of systemic cytokine levels. Additionally, inhibition of STIM1 signaling prevented receptor-interacting protein 3-dependent (RIP3-dependent) EC death. Remarkably, BTP2, a small-molecule calcium release-activated calcium (CRAC) channel blocker administered after insult, halted LPS-induced vascular leakage and pulmonary edema. These results indicate that ROS-driven Ca2+ signaling promotes vascular barrier dysfunction and that the SOCe machinery may provide crucial therapeutic targets to limit sepsis-induced ALI.
Figures








Comment in
-
Calcium flux and endothelial dysfunction during acute lung injury: a STIMulating target for therapy.J Clin Invest. 2013 Mar;123(3):1015-8. doi: 10.1172/JCI68093. Epub 2013 Feb 22. J Clin Invest. 2013. PMID: 23434597 Free PMC article.
Similar articles
-
STIM1 Regulates Endothelial Calcium Overload and Cytokine Upregulation During Sepsis.J Surg Res. 2021 Jul;263:236-244. doi: 10.1016/j.jss.2020.12.016. Epub 2021 Mar 10. J Surg Res. 2021. PMID: 33713955
-
Calcium flux and endothelial dysfunction during acute lung injury: a STIMulating target for therapy.J Clin Invest. 2013 Mar;123(3):1015-8. doi: 10.1172/JCI68093. Epub 2013 Feb 22. J Clin Invest. 2013. PMID: 23434597 Free PMC article.
-
Nox2-dependent glutathionylation of endothelial NOS leads to uncoupled superoxide production and endothelial barrier dysfunction in acute lung injury.Am J Physiol Lung Cell Mol Physiol. 2014 Dec 15;307(12):L987-97. doi: 10.1152/ajplung.00063.2014. Epub 2014 Oct 17. Am J Physiol Lung Cell Mol Physiol. 2014. PMID: 25326583 Free PMC article.
-
NADPH Oxidase-2 and Atherothrombosis: Insight From Chronic Granulomatous Disease.Arterioscler Thromb Vasc Biol. 2017 Feb;37(2):218-225. doi: 10.1161/ATVBAHA.116.308351. Epub 2016 Dec 8. Arterioscler Thromb Vasc Biol. 2017. PMID: 27932349 Review.
-
The regulatory and modulatory roles of TRP family channels in malignant tumors and relevant therapeutic strategies.Acta Pharm Sin B. 2022 Apr;12(4):1761-1780. doi: 10.1016/j.apsb.2021.11.001. Epub 2021 Nov 5. Acta Pharm Sin B. 2022. PMID: 35847486 Free PMC article. Review.
Cited by
-
Mesenchymal stem cells activate Notch signaling to induce regulatory dendritic cells in LPS-induced acute lung injury.J Transl Med. 2020 Jun 16;18(1):241. doi: 10.1186/s12967-020-02410-z. J Transl Med. 2020. PMID: 32546185 Free PMC article.
-
Homer1a attenuates glutamate-induced oxidative injury in HT-22 cells through regulation of store-operated calcium entry.Sci Rep. 2016 Sep 29;6:33975. doi: 10.1038/srep33975. Sci Rep. 2016. PMID: 27681296 Free PMC article.
-
The role of Ca2+/NFAT in Dysfunction and Inflammation of Human Coronary Endothelial Cells induced by Sera from patients with Kawasaki disease.Sci Rep. 2020 Mar 13;10(1):4706. doi: 10.1038/s41598-020-61667-y. Sci Rep. 2020. PMID: 32170198 Free PMC article.
-
Oxidative stress increases pulmonary vascular permeability in diabetic rats through activation of transient receptor potential melastatin 2 channels.Microcirculation. 2014 Nov;21(8):754-60. doi: 10.1111/micc.12158. Microcirculation. 2014. PMID: 25059284 Free PMC article.
-
Calcium Signals during SARS-CoV-2 Infection: Assessing the Potential of Emerging Therapies.Cells. 2022 Jan 12;11(2):253. doi: 10.3390/cells11020253. Cells. 2022. PMID: 35053369 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
- R21HL109920-01/HL/NHLBI NIH HHS/United States
- R01 HL108910/HL/NHLBI NIH HHS/United States
- R01 HL077288/HL/NHLBI NIH HHS/United States
- P01 HL080101/HL/NHLBI NIH HHS/United States
- HL086699/HL/NHLBI NIH HHS/United States
- R01 HL117654/HL/NHLBI NIH HHS/United States
- F32 HL105066/HL/NHLBI NIH HHS/United States
- S10 RR027327/RR/NCRR NIH HHS/United States
- R01 AI058173/AI/NIAID NIH HHS/United States
- R21 HL109920/HL/NHLBI NIH HHS/United States
- R01 HL086699/HL/NHLBI NIH HHS/United States
- R01 HL110764/HL/NHLBI NIH HHS/United States
- 1S10RR027327-01/RR/NCRR NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
