

Lovastatin corrects excess protein synthesis and prevents epileptogenesis in a mouse model of fragile X syndrome
- PMID: 23352161
- PMCID: PMC3597444
- DOI: 10.1016/j.neuron.2012.01.034
Lovastatin corrects excess protein synthesis and prevents epileptogenesis in a mouse model of fragile X syndrome
Abstract
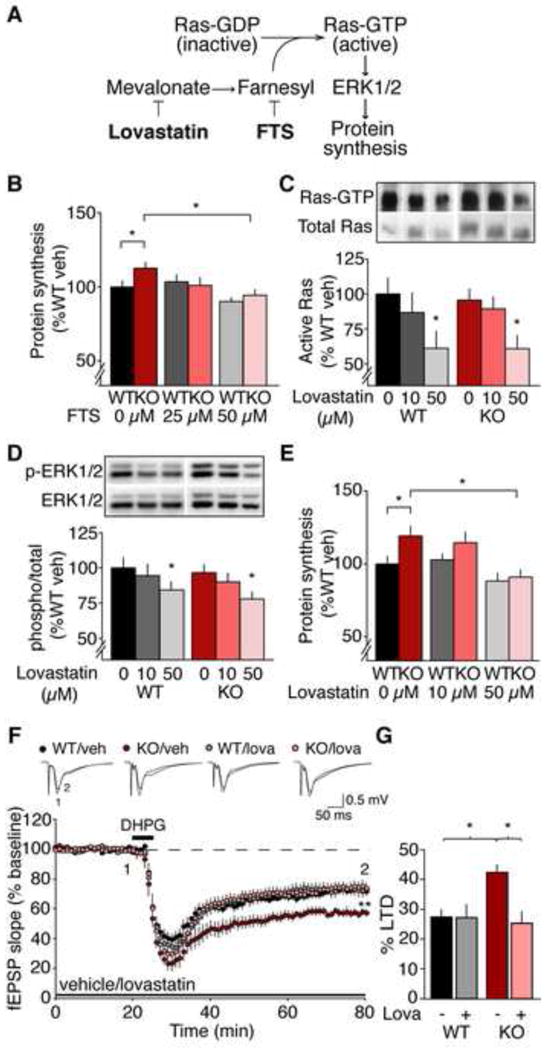
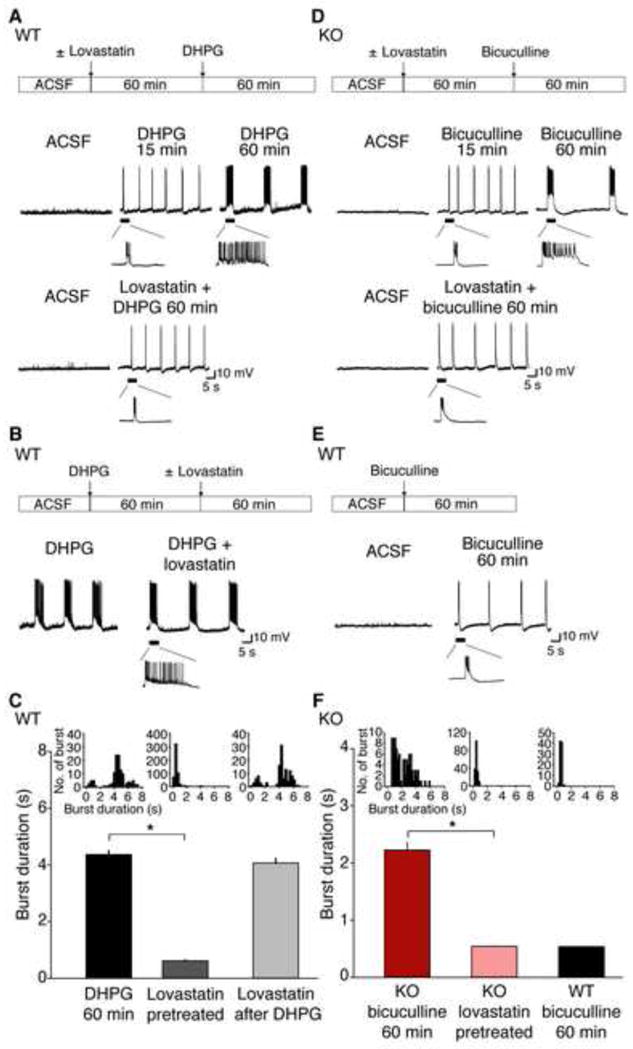
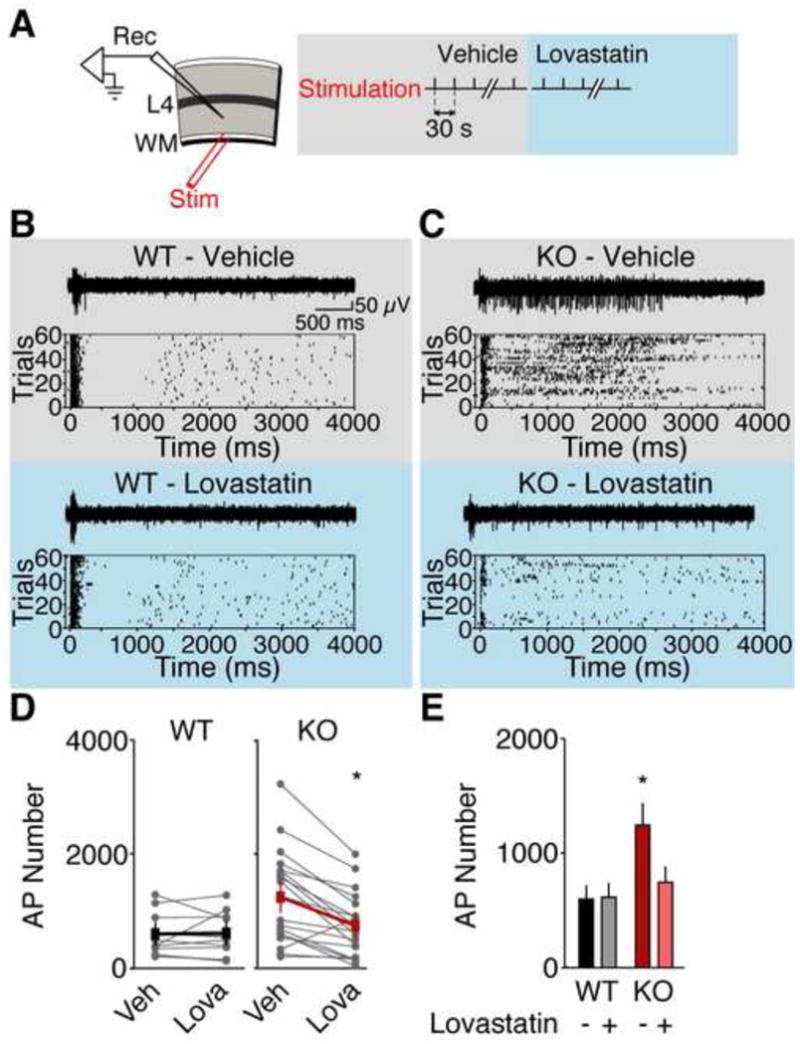
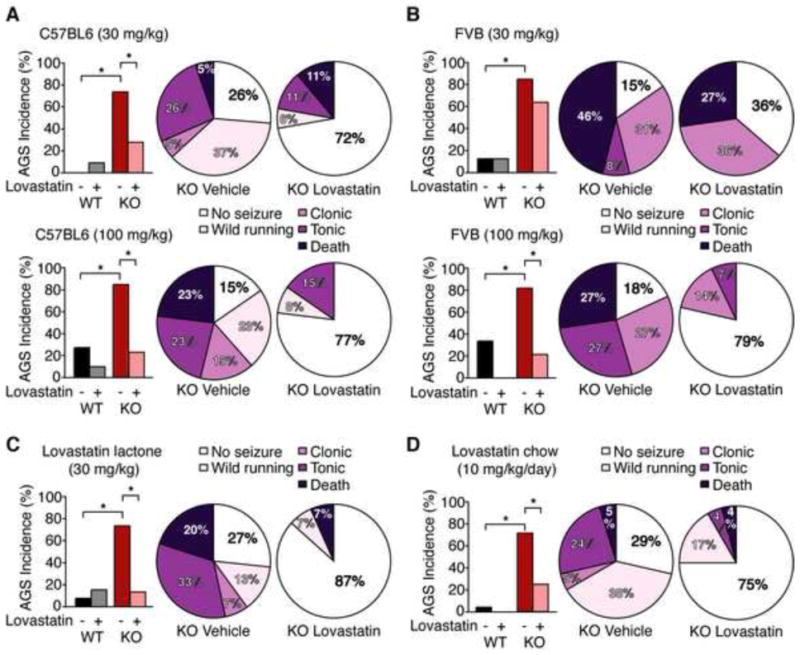
Many neuropsychiatric symptoms of fragile X syndrome (FXS) are believed to be a consequence of altered regulation of protein synthesis at synapses. We discovered that lovastatin, a drug that is widely prescribed for the treatment of high cholesterol, can correct excess hippocampal protein synthesis in the mouse model of FXS and can prevent one of the robust functional consequences of increased protein synthesis in FXS, epileptogenesis. These data suggest that lovastatin is potentially disease modifying and could be a viable prophylactic treatment for epileptogenesis in FXS.
Copyright © 2013 Elsevier Inc. All rights reserved.
Conflict of interest statement
Figures
Comment in
-
Fragile X syndrome therapeutics: translation, meet translational medicine.Neuron. 2013 Jan 23;77(2):212-3. doi: 10.1016/j.neuron.2013.01.009. Neuron. 2013. PMID: 23352156
References
-
- Bear MF, Huber KM, Warren ST. The mGluR theory of fragile X mental retardation. Trends Neurosci. 2004;27:370–377. - PubMed
-
- Berry-Kravis E. Epilepsy in fragile X syndrome. Dev Med Child Neurol. 2002;44:724–728. - PubMed
-
- Choi CH, Schoenfeld BP, Bell AJ, Hinchey P, Kollaros M, Gertner MJ, Woo NH, Tranfaglia MR, Bear MF, Zukin RS, et al. Pharmacological reversal of synaptic plasticity deficits in the mouse model of fragile X syndrome by group II mGluR antagonist or lithium treatment. Brain Res. 2011;1380:106–119. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
