

Identification of early replicating fragile sites that contribute to genome instability
- PMID: 23352430
- PMCID: PMC3629730
- DOI: 10.1016/j.cell.2013.01.006
Identification of early replicating fragile sites that contribute to genome instability
Abstract
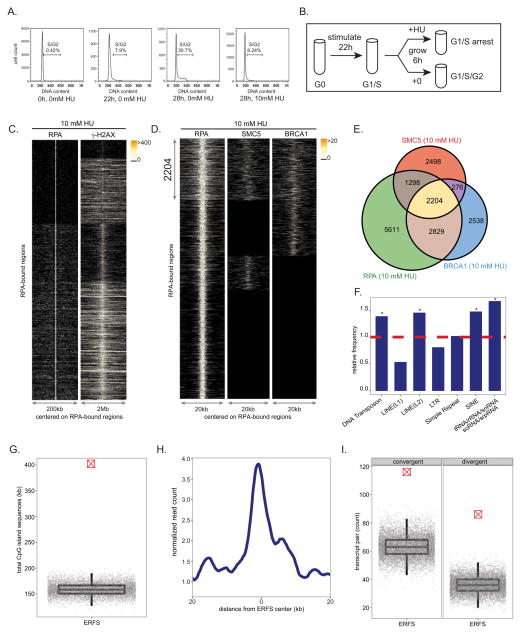
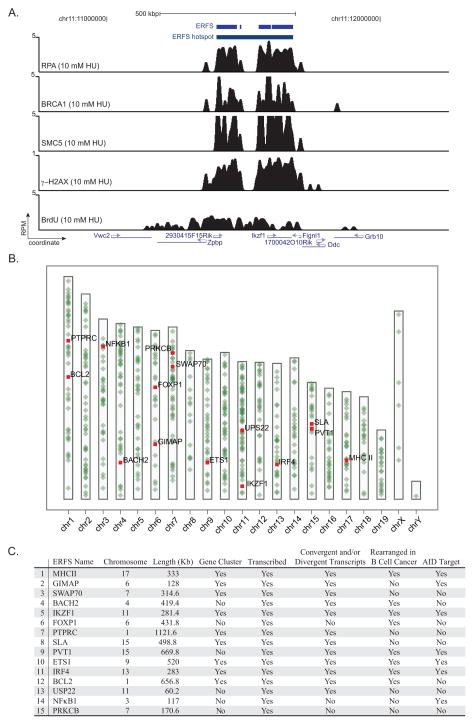
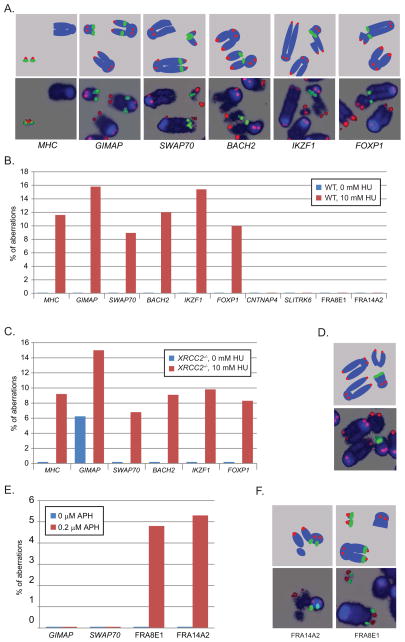
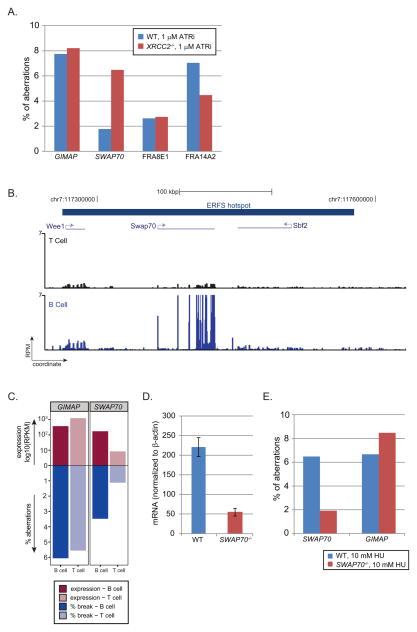
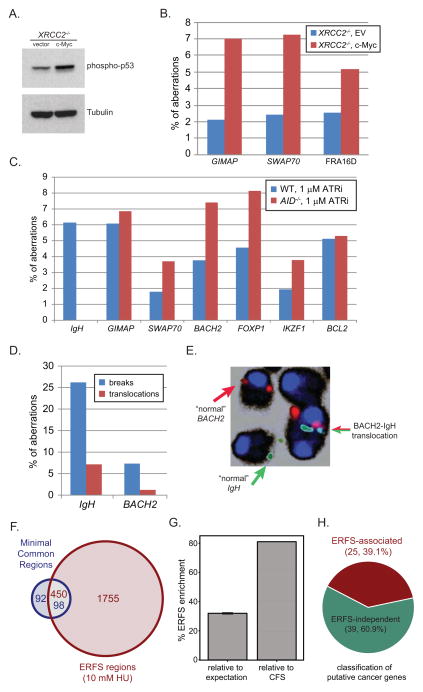
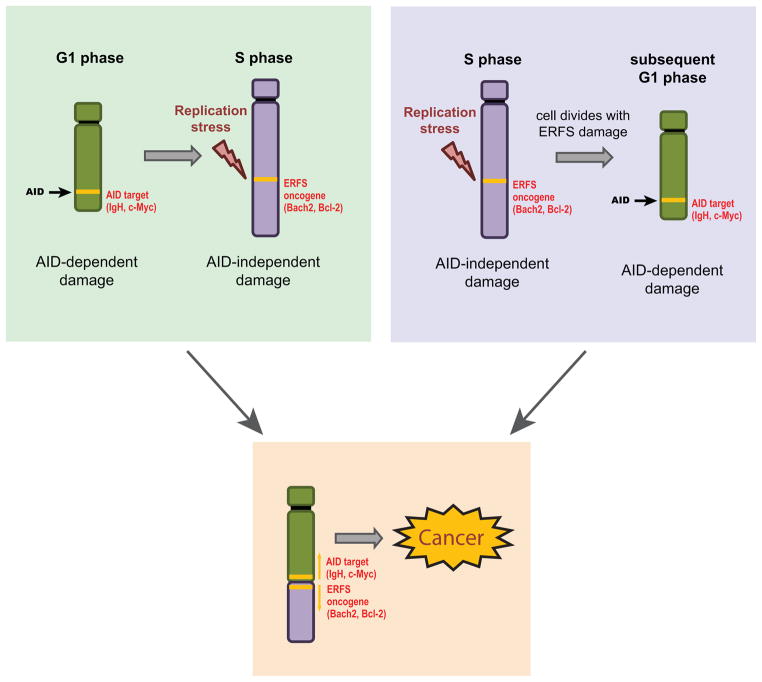
DNA double-strand breaks (DSBs) in B lymphocytes arise stochastically during replication or as a result of targeted DNA damage by activation-induced cytidine deaminase (AID). Here we identify recurrent, early replicating, and AID-independent DNA lesions, termed early replication fragile sites (ERFSs), by genome-wide localization of DNA repair proteins in B cells subjected to replication stress. ERFSs colocalize with highly expressed gene clusters and are enriched for repetitive elements and CpG dinucleotides. Although distinct from late-replicating common fragile sites (CFS), the stability of ERFSs and CFSs is similarly dependent on the replication-stress response kinase ATR. ERFSs break spontaneously during replication, but their fragility is increased by hydroxyurea, ATR inhibition, or deregulated c-Myc expression. Moreover, greater than 50% of recurrent amplifications/deletions in human diffuse large B cell lymphoma map to ERFSs. In summary, we have identified a source of spontaneous DNA lesions that drives instability at preferred genomic sites.
Copyright © 2013 Elsevier Inc. All rights reserved.
Figures
Comment in
-
DNA damage: In at the beginning.Nat Rev Cancer. 2013 Mar;13(3):147. doi: 10.1038/nrc3472. Epub 2013 Feb 7. Nat Rev Cancer. 2013. PMID: 23388616 No abstract available.
-
Breaking news on fragile sites in cancer.Cancer Cell. 2013 Feb 11;23(2):137-9. doi: 10.1016/j.ccr.2013.01.017. Cancer Cell. 2013. PMID: 23410970
-
Early replication fragile sites may underlie gene rearrangements.Cancer Discov. 2013 Mar;3(3):OF11. doi: 10.1158/2159-8290.CD-RW2013-026. Epub 2013 Jan 31. Cancer Discov. 2013. PMID: 23475881
References
-
- Bartek J, Bartkova J, Lukas J. DNA damage signalling guards against activated oncogenes and tumour progression. Oncogene. 2007;26:7773–7779. - PubMed
-
- Borggrefe T, Keshavarzi S, Gross B, Wabl M, Jessberger R. Impaired IgE response in SWAP-70-deficient mice. European journal of immunology. 2001;31:2467–2475. - PubMed
Publication types
MeSH terms
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
