

Helicase-mediated changes in RNA structure at the single-molecule level
- PMID: 23353571
- PMCID: PMC3590230
- DOI: 10.4161/rna.23507
Helicase-mediated changes in RNA structure at the single-molecule level
Abstract
RNA helicases are a diverse group of RNA-dependent ATPases known to play a large number of biological roles inside the cell, such as RNA unwinding, remodeling, export and degradation. Understanding how helicases mediate changes in RNA structure is therefore of fundamental interest. The advent of single-molecule spectroscopic techniques has unveiled with unprecedented detail the interplay of RNA helicases with their substrates. In this review, we describe the characterization of helicase-RNA interactions by single-molecule approaches. State-of-the-art techniques are presented, followed by a discussion of recent advancements in this exciting field.
Keywords: AFM; DEAD-box; FRET; PIFE; RNA folding; RNA helicase; optical tweezers; single-molecule spectroscopy.
Figures
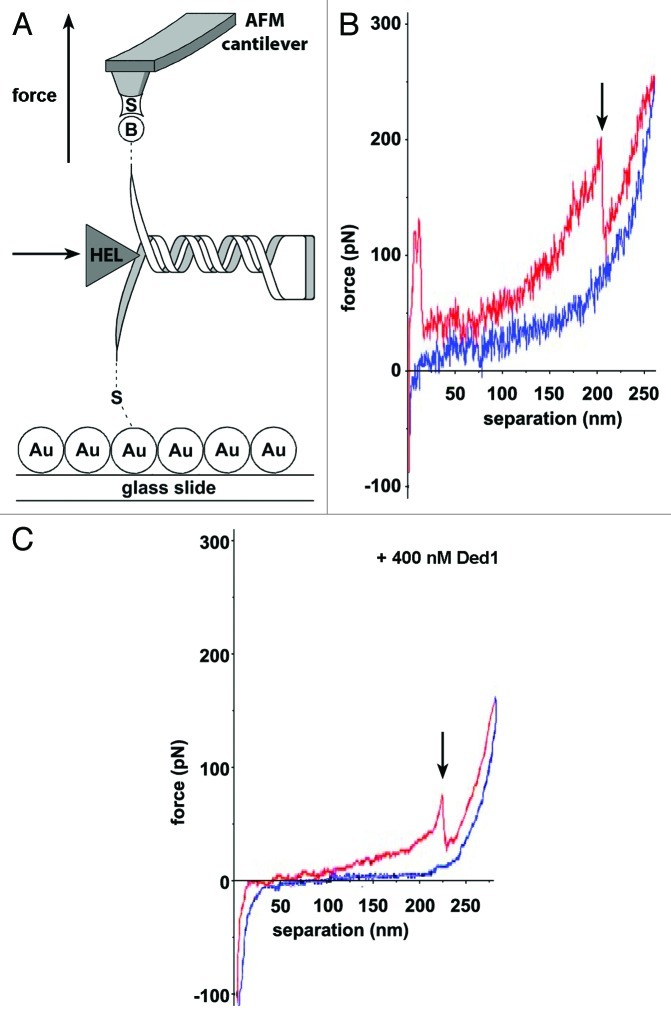
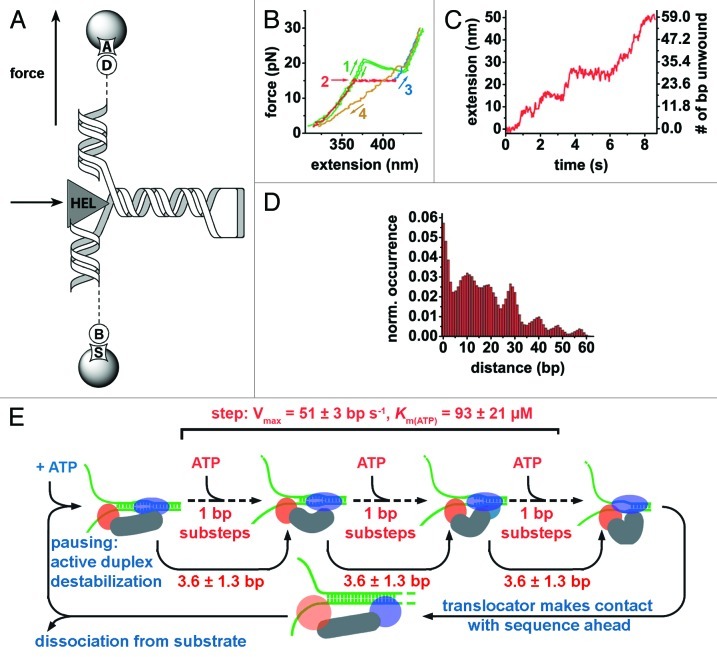
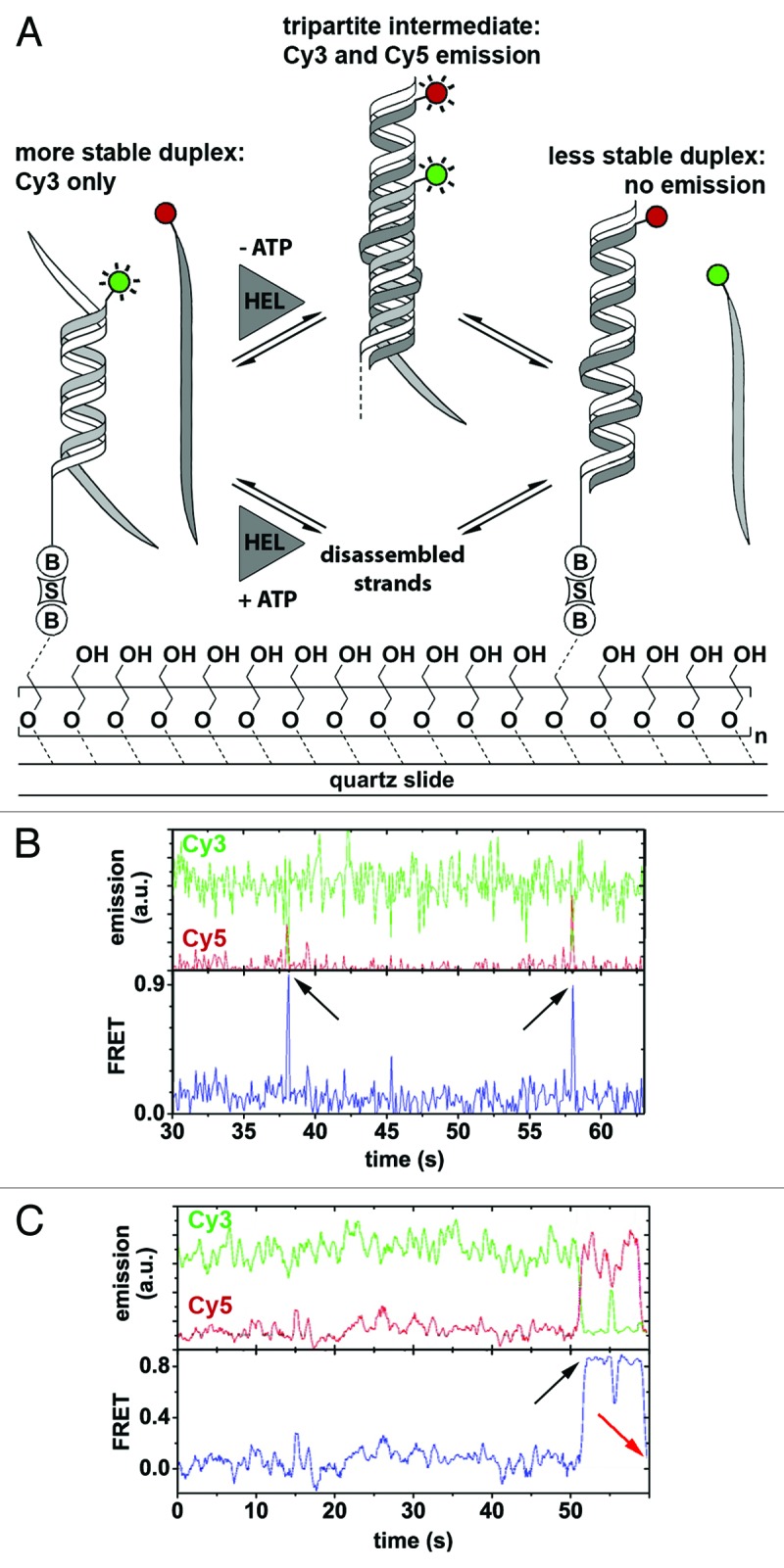
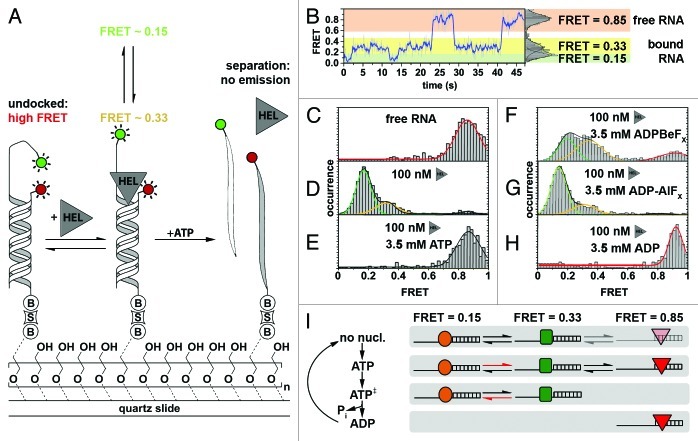
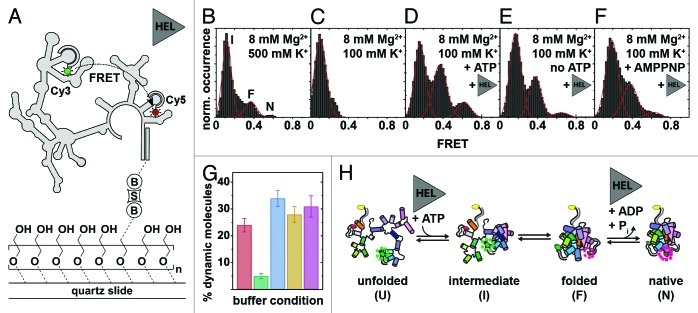
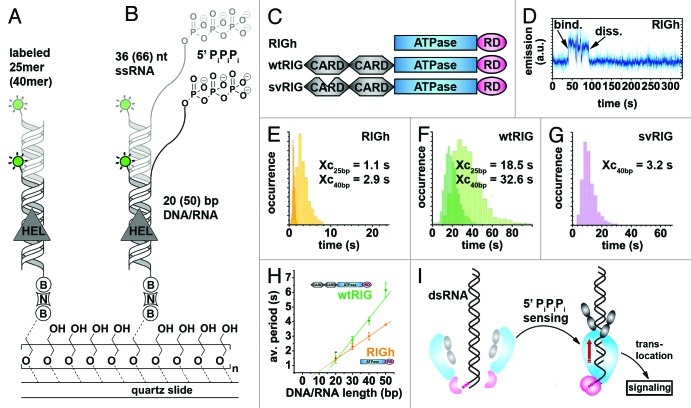
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous