

A large-scale evaluation of computational protein function prediction
- PMID: 23353650
- PMCID: PMC3584181
- DOI: 10.1038/nmeth.2340
A large-scale evaluation of computational protein function prediction
Abstract
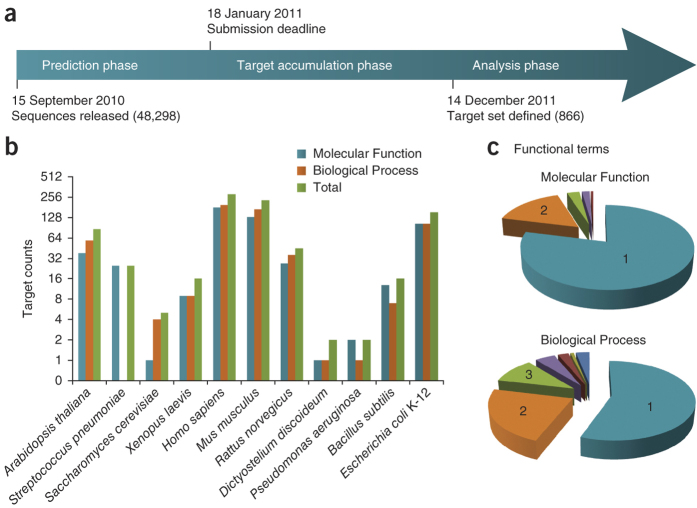
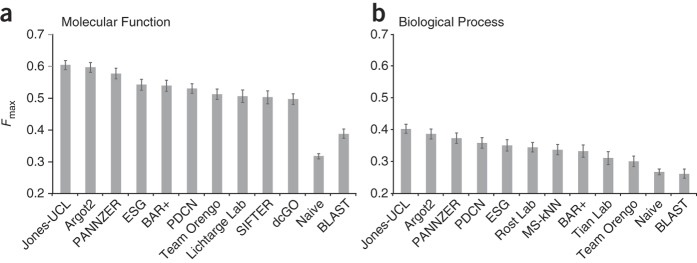
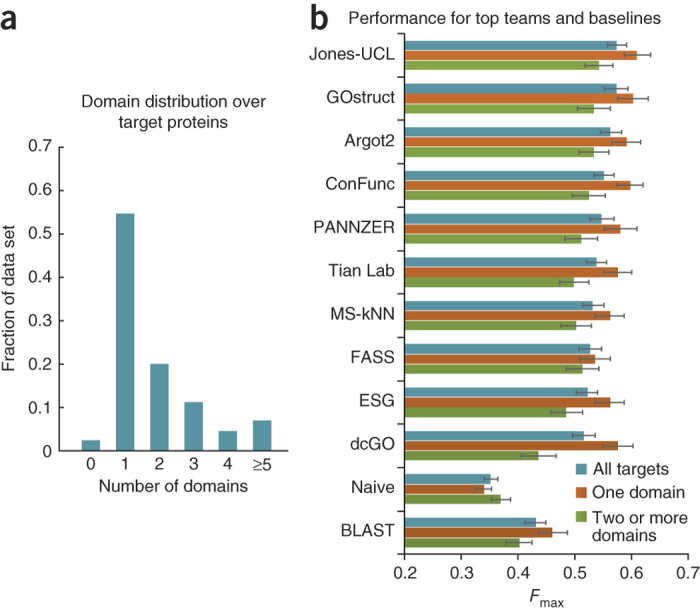
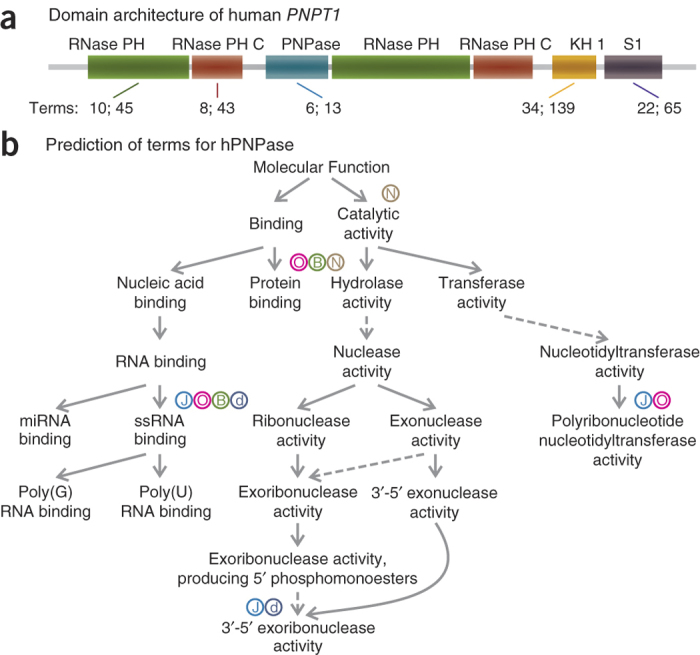
Automated annotation of protein function is challenging. As the number of sequenced genomes rapidly grows, the overwhelming majority of protein products can only be annotated computationally. If computational predictions are to be relied upon, it is crucial that the accuracy of these methods be high. Here we report the results from the first large-scale community-based critical assessment of protein function annotation (CAFA) experiment. Fifty-four methods representing the state of the art for protein function prediction were evaluated on a target set of 866 proteins from 11 organisms. Two findings stand out: (i) today's best protein function prediction algorithms substantially outperform widely used first-generation methods, with large gains on all types of targets; and (ii) although the top methods perform well enough to guide experiments, there is considerable need for improvement of currently available tools.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
- GM097528/GM/NIGMS NIH HHS/United States
- T32 HG000047/HG/NHGRI NIH HHS/United States
- GM093123/GM/NIGMS NIH HHS/United States
- R01 GM066099/GM/NIGMS NIH HHS/United States
- BB/F020481/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BB/F00964X/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- R01 LM009722/LM/NLM NIH HHS/United States
- R01 GM060595/GM/NIGMS NIH HHS/United States
- BB/K004131/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BB/H02364X/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- R01 GM093123/GM/NIGMS NIH HHS/United States
- GM075004/GM/NIGMS NIH HHS/United States
- R01 GM071749/GM/NIGMS NIH HHS/United States
- R01 GM079656/GM/NIGMS NIH HHS/United States
- GM079656/GM/NIGMS NIH HHS/United States
- BB/G022771/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- LM009722/LM/NLM NIH HHS/United States
- HG004028/HG/NHGRI NIH HHS/United States
- R13 HG006079-01A1/HG/NHGRI NIH HHS/United States
- U54 HG004028/HG/NHGRI NIH HHS/United States
- GM066099/GM/NIGMS NIH HHS/United States
- LM00945102/LM/NLM NIH HHS/United States
- R13 HG006079/HG/NHGRI NIH HHS/United States
- R01 GM097528/GM/NIGMS NIH HHS/United States
- R01 GM075004/GM/NIGMS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
