

Cerulein-induced chronic pancreatitis does not require intra-acinar activation of trypsinogen in mice
- PMID: 23354015
- PMCID: PMC3928043
- DOI: 10.1053/j.gastro.2013.01.041
Cerulein-induced chronic pancreatitis does not require intra-acinar activation of trypsinogen in mice
Abstract
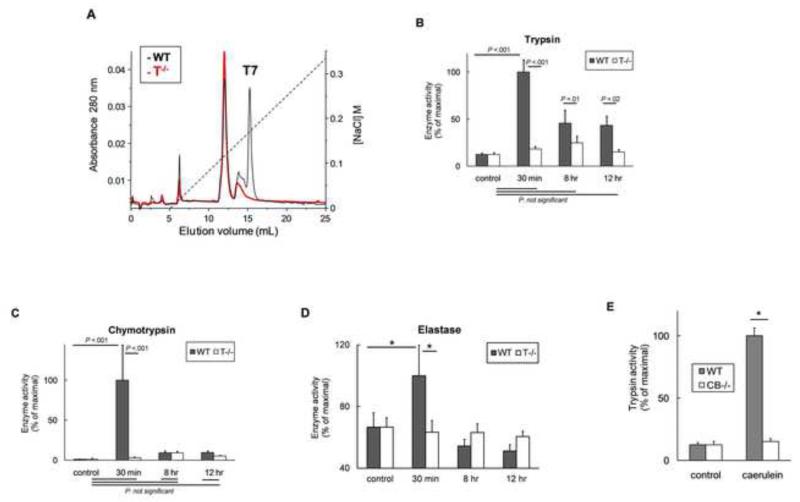
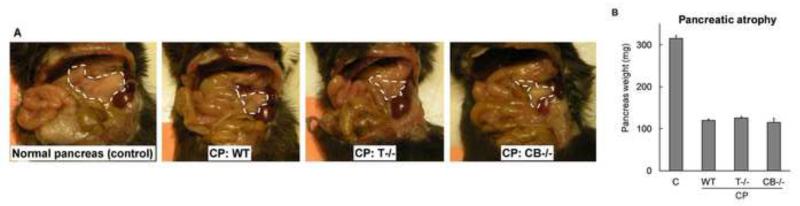
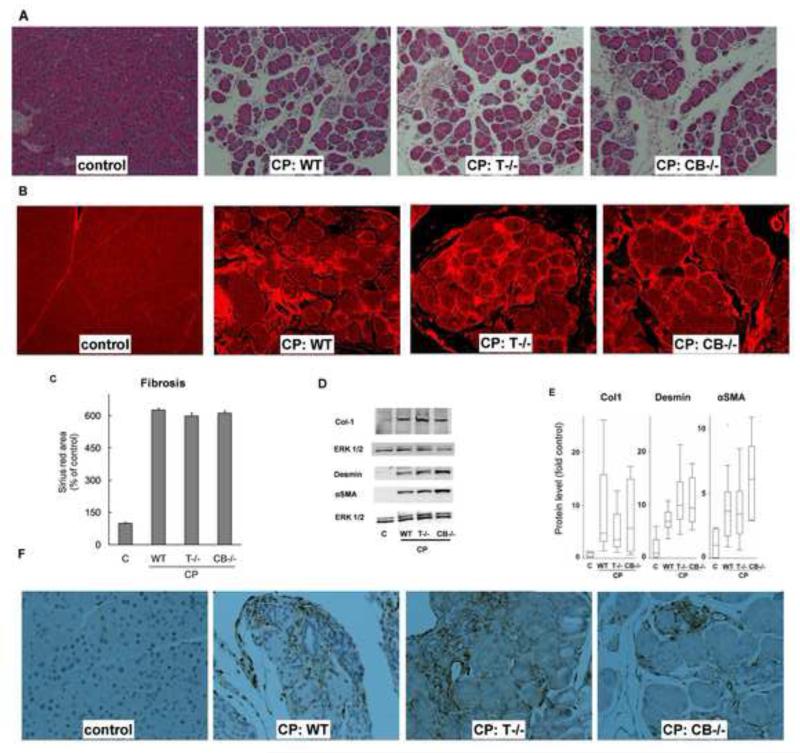
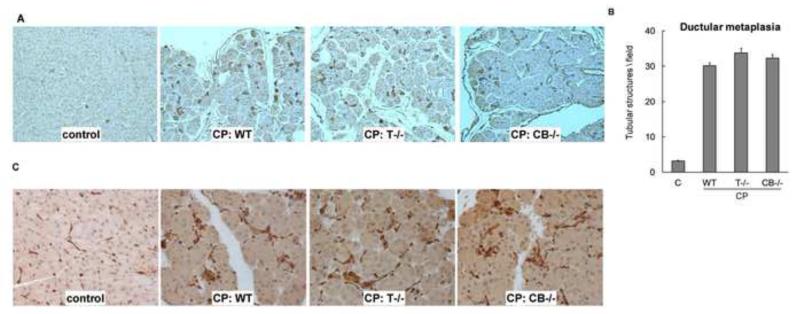
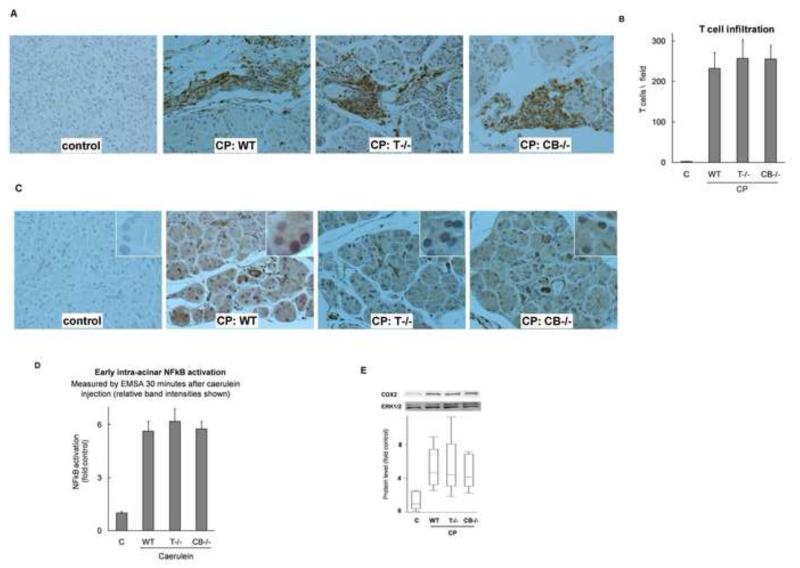
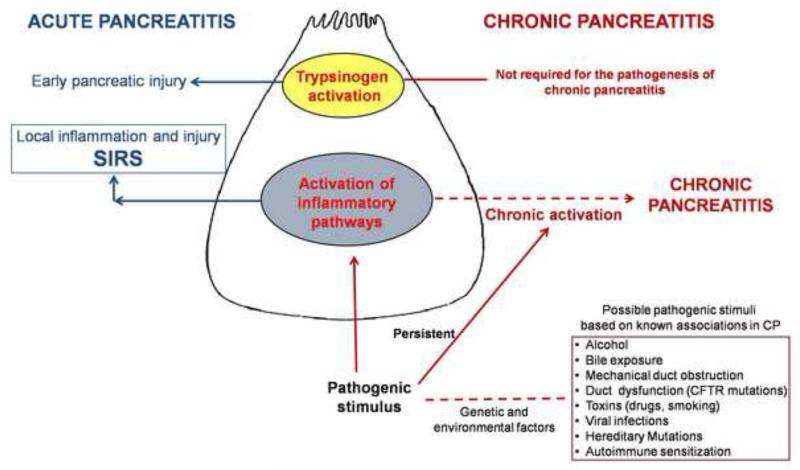
Background & aims: Premature activation of trypsinogen activation can cause pancreatic injury and has been associated with chronic pancreatitis (CP). Mice that lack intra-acinar activation of trypsinogen, such as trypsinogen-7-null (T(-/-)) and cathepsin B-null (CB(-/-)) mice, have been used to study trypsin-independent processes of CP development. We compared histologic features and inflammatory responses of pancreatic tissues from these mice with those from wild-type mice after the development of CP.
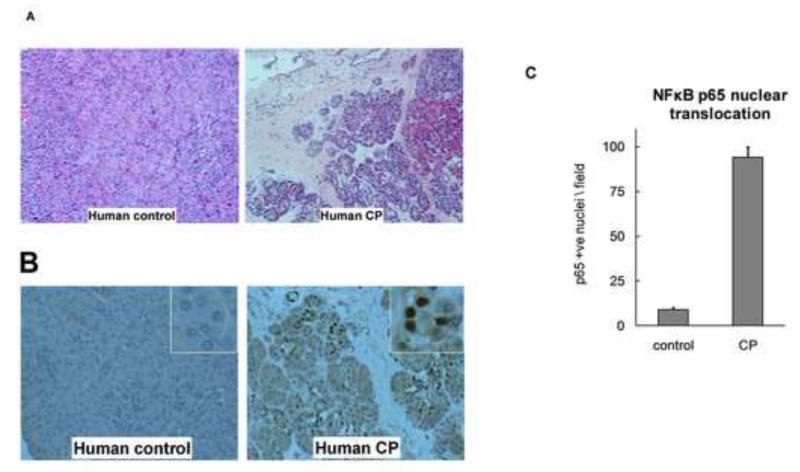
Methods: CP was induced in wild-type, T(-/-), and CB(-/-) mice by twice-weekly induction of acute pancreatitis for 10 weeks; acute pancreatitis was induced by hourly intraperitoneal injections of cerulein (50 μg/kg × 6). Pancreatic samples were collected and evaluated by histologic and immunohistochemical analyses. Normal human pancreas samples, obtained from the islet transplant program at the University of Minnesota, were used as controls and CP samples were obtained from surgical resections.
Results: Compared with pancreatic tissues from wild-type mice, those from T(-/-) and CB(-/-) mice had similar levels of atrophy, histomorphologic features of CP, and chronic inflammation. All samples had comparable intra-acinar activation of nuclear factor (NF)-κB, a transcription factor that regulates the inflammatory response, immediately after injection of cerulein. Pancreatic tissue samples from patients with CP had increased activation of NF-κB (based on nuclear translocation of p65 in acinar cells) compared with controls.
Conclusions: Induction of CP in mice by cerulein injection does not require intra-acinar activation of trypsinogen. Pancreatic acinar cells of patients with CP have increased levels of NF-κB activation compared with controls; regulation of the inflammatory response by this transcription factor might be involved in the pathogenesis of CP.
Copyright © 2013 AGA Institute. Published by Elsevier Inc. All rights reserved.
Figures
Similar articles
-
Cathepsin B-Mediated Activation of Trypsinogen in Endocytosing Macrophages Increases Severity of Pancreatitis in Mice.Gastroenterology. 2018 Feb;154(3):704-718.e10. doi: 10.1053/j.gastro.2017.10.018. Epub 2018 Jan 10. Gastroenterology. 2018. PMID: 29079517 Free PMC article.
-
Intra-acinar trypsinogen activation mediates early stages of pancreatic injury but not inflammation in mice with acute pancreatitis.Gastroenterology. 2011 Dec;141(6):2210-2217.e2. doi: 10.1053/j.gastro.2011.08.033. Epub 2011 Aug 27. Gastroenterology. 2011. PMID: 21875495 Free PMC article.
-
Endoplasmic reticulum stress is chronically activated in chronic pancreatitis.J Biol Chem. 2014 Oct 3;289(40):27551-61. doi: 10.1074/jbc.M113.528174. Epub 2014 Jul 30. J Biol Chem. 2014. PMID: 25077966 Free PMC article.
-
Early Intra-Acinar Events in Pathogenesis of Pancreatitis.Gastroenterology. 2019 May;156(7):1979-1993. doi: 10.1053/j.gastro.2019.01.268. Epub 2019 Feb 15. Gastroenterology. 2019. PMID: 30776339 Review.
-
Pathogenic mechanisms of acute pancreatitis.Curr Opin Gastroenterol. 2012 Sep;28(5):507-15. doi: 10.1097/MOG.0b013e3283567f52. Curr Opin Gastroenterol. 2012. PMID: 22885948 Free PMC article. Review.
Cited by
-
Sphingosine 1-phosphate receptor 2 mediated early stages of pancreatic and systemic inflammatory responses via NF-kappa B activation in acute pancreatitis.Cell Commun Signal. 2022 Oct 13;20(1):157. doi: 10.1186/s12964-022-00971-8. Cell Commun Signal. 2022. PMID: 36229875 Free PMC article.
-
Role of leucine-rich repeat kinase 2 in severe acute pancreatitis.Front Immunol. 2024 Feb 19;15:1364839. doi: 10.3389/fimmu.2024.1364839. eCollection 2024. Front Immunol. 2024. PMID: 38440723 Free PMC article. Review.
-
Pancreatic Disorders.Pediatr Clin North Am. 2017 Jun;64(3):685-706. doi: 10.1016/j.pcl.2017.01.010. Pediatr Clin North Am. 2017. PMID: 28502446 Free PMC article. Review.
-
Pancreatic Ubap2 deletion regulates glucose tolerance, inflammation, and protection from cerulein-induced pancreatitis.Cancer Lett. 2023 Dec 1;578:216455. doi: 10.1016/j.canlet.2023.216455. Epub 2023 Oct 19. Cancer Lett. 2023. PMID: 37865160 Free PMC article.
-
Animal Models: Challenges and Opportunities to Determine Optimal Experimental Models of Pancreatitis and Pancreatic Cancer.Pancreas. 2019 Jul;48(6):759-779. doi: 10.1097/MPA.0000000000001335. Pancreas. 2019. PMID: 31206467 Free PMC article. Review.
References
-
- Barrett KE, et al. Barrett KE, Barman SM, Boitano S, Brooks H, editors. Protein Digestion. Ganong’s Review of Medical Physiology. (23 ed) 2010 p. http://www.accessmedicine.com/content.aspx?aID=5242356.
-
- Chiari H. ÜberdieSelbstverdauung des menschlichenPankreas. ZeitschriftfürHeilkunde. 1896;17:69–96.
-
- Saluja AK, Lerch MM, et al. Why does pancreatic overstimulation cause pancreatitis? Annu Rev Physiol. 2007;69:249–69. - PubMed
-
- Hofbauer B, Saluja AK, et al. Intra-acinar cell activation of trypsinogen during caerulein-induced pancreatitis in rats. Am J Physiol. 1998;275(2 Pt 1):G352–62. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
