

Missense mutations in β-1,3-N-acetylglucosaminyltransferase 1 (B3GNT1) cause Walker-Warburg syndrome
- PMID: 23359570
- PMCID: PMC3613162
- DOI: 10.1093/hmg/ddt021
Missense mutations in β-1,3-N-acetylglucosaminyltransferase 1 (B3GNT1) cause Walker-Warburg syndrome
Abstract
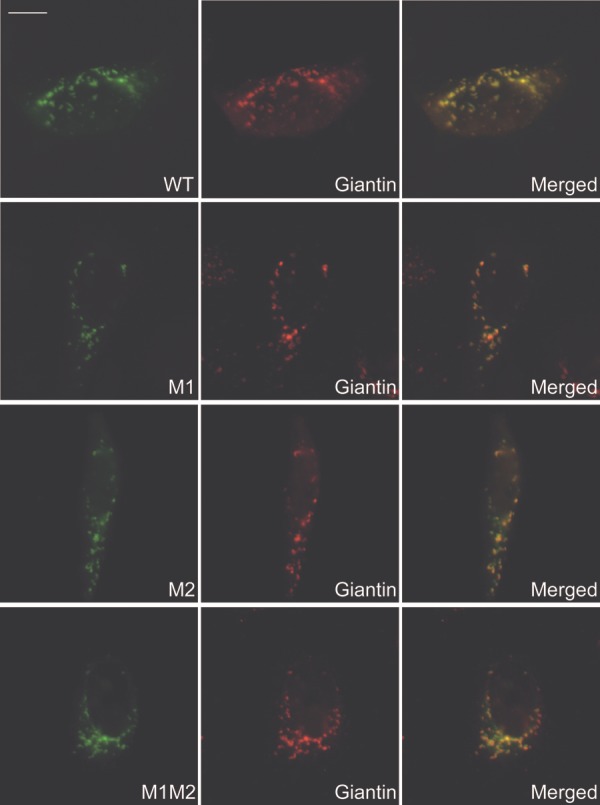
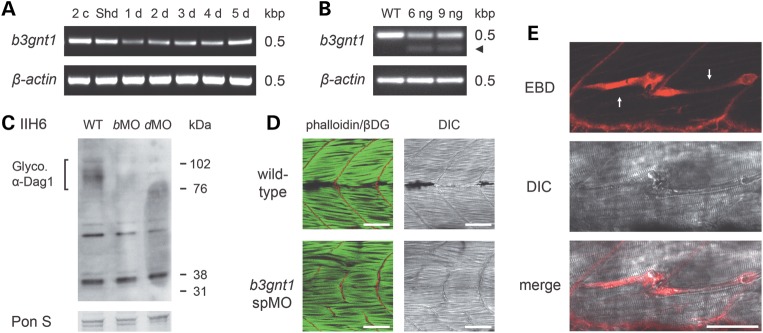
Several known or putative glycosyltransferases are required for the synthesis of laminin-binding glycans on alpha-dystroglycan (αDG), including POMT1, POMT2, POMGnT1, LARGE, Fukutin, FKRP, ISPD and GTDC2. Mutations in these glycosyltransferase genes result in defective αDG glycosylation and reduced ligand binding by αDG causing a clinically heterogeneous group of congenital muscular dystrophies, commonly referred to as dystroglycanopathies. The most severe clinical form, Walker-Warburg syndrome (WWS), is characterized by congenital muscular dystrophy and severe neurological and ophthalmological defects. Here, we report two homozygous missense mutations in the β-1,3-N-acetylglucosaminyltransferase 1 (B3GNT1) gene in a family affected with WWS. Functional studies confirmed the pathogenicity of the mutations. First, expression of wild-type but not mutant B3GNT1 in human prostate cancer (PC3) cells led to increased levels of αDG glycosylation. Second, morpholino knockdown of the zebrafish b3gnt1 orthologue caused characteristic muscular defects and reduced αDG glycosylation. These functional studies identify an important role of B3GNT1 in the synthesis of the uncharacterized laminin-binding glycan of αDG and implicate B3GNT1 as a novel causative gene for WWS.
Figures



Similar articles
-
Refining genotype phenotype correlations in muscular dystrophies with defective glycosylation of dystroglycan.Brain. 2007 Oct;130(Pt 10):2725-35. doi: 10.1093/brain/awm212. Epub 2007 Sep 18. Brain. 2007. PMID: 17878207
-
A truncating mutation in B3GNT1 causes severe Walker-Warburg syndrome.Neurogenetics. 2013 Nov;14(3-4):243-5. doi: 10.1007/s10048-013-0367-8. Epub 2013 Jul 23. Neurogenetics. 2013. PMID: 23877401
-
160 kb deletion in ISPD unmasking a recessive mutation in a patient with Walker-Warburg syndrome.Eur J Med Genet. 2013 Dec;56(12):689-94. doi: 10.1016/j.ejmg.2013.09.014. Epub 2013 Oct 10. Eur J Med Genet. 2013. PMID: 24120487
-
[Recent Advances in α-dystroglycanopathy].Brain Nerve. 2011 Nov;63(11):1189-95. Brain Nerve. 2011. PMID: 22068471 Review. Japanese.
-
Glyc-O-genetics of Walker-Warburg syndrome.Clin Genet. 2005 Apr;67(4):281-9. doi: 10.1111/j.1399-0004.2004.00368.x. Clin Genet. 2005. PMID: 15733261 Review.
Cited by
-
The potential of sarcospan in adhesion complex replacement therapeutics for the treatment of muscular dystrophy.FEBS J. 2013 Sep;280(17):4210-29. doi: 10.1111/febs.12295. Epub 2013 May 13. FEBS J. 2013. PMID: 23601082 Free PMC article. Review.
-
Clear differences in cerebrospinal fluid proteome between women with chronic widespread pain and healthy women - a multivariate explorative cross-sectional study.J Pain Res. 2017 Mar 13;10:575-590. doi: 10.2147/JPR.S125667. eCollection 2017. J Pain Res. 2017. PMID: 28331360 Free PMC article.
-
The transgenic expression of LARGE exacerbates the muscle phenotype of dystroglycanopathy mice.Hum Mol Genet. 2014 Apr 1;23(7):1842-55. doi: 10.1093/hmg/ddt577. Epub 2013 Nov 13. Hum Mol Genet. 2014. PMID: 24234655 Free PMC article.
-
Biallelic Mutations in TMTC3, Encoding a Transmembrane and TPR-Containing Protein, Lead to Cobblestone Lissencephaly.Am J Hum Genet. 2016 Nov 3;99(5):1181-1189. doi: 10.1016/j.ajhg.2016.09.007. Epub 2016 Oct 20. Am J Hum Genet. 2016. PMID: 27773428 Free PMC article.
-
The roles of dystroglycan in the nervous system: insights from animal models of muscular dystrophy.Dis Model Mech. 2018 Dec 19;11(12):dmm035931. doi: 10.1242/dmm.035931. Dis Model Mech. 2018. PMID: 30578246 Free PMC article. Review.
References
-
- Michele D.E., Barresi R., Kanagawa M., Saito F., Cohn R.D., Satz J.S., Dollar J., Nishino I., Kelley R.I., Somer H., et al. Post-translational disruption of dystroglycan-ligand interactions in congenital muscular dystrophies. Nature. 2002;418:417–422. - PubMed
-
- Moore S.A., Saito F., Chen J., Michele D.E., Henry M.D., Messing A., Cohn R.D., Ross-Barta S.E., Westra S., Williamson R.A., et al. Deletion of brain dystroglycan recapitulates aspects of congenital muscular dystrophy. Nature. 2002;418:422–425. - PubMed
-
- van Reeuwijk J., Brunner H.G., van Bokhoven H. Glyc-O-genetics of Walker–Warburg syndrome. Clin. Genet. 2005;67:281–289. - PubMed
-
- Brockington M., Blake D.J., Prandini P., Brown S.C., Torelli S., Benson M.A., Ponting C.P., Estournet B., Romero N.B., Mercuri E., et al. Mutations in the fukutin-related protein gene (FKRP) cause a form of congenital muscular dystrophy with secondary laminin α2 deficiency and abnormal glycosylation of α-dystroglycan. Am. J. Hum. Genet. 2001;69:1198–1209. - PMC - PubMed
-
- Henry M.D., Campbell K.P. Dystroglycan: an extracellular matrix receptor linked to the cytoskeleton. Curr. Opin. Cell Biol. 1996;8:625–631. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
