

Genotype-phenotype correlation in 1,507 families with congenital adrenal hyperplasia owing to 21-hydroxylase deficiency
- PMID: 23359698
- PMCID: PMC3574953
- DOI: 10.1073/pnas.1300057110
Genotype-phenotype correlation in 1,507 families with congenital adrenal hyperplasia owing to 21-hydroxylase deficiency
Abstract
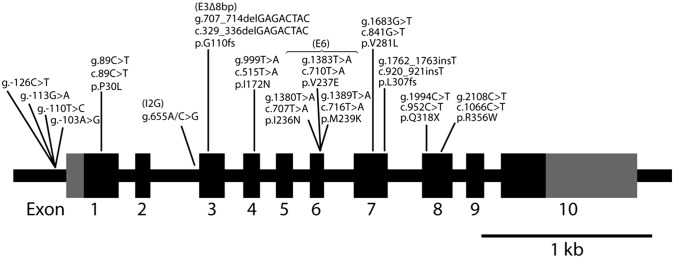
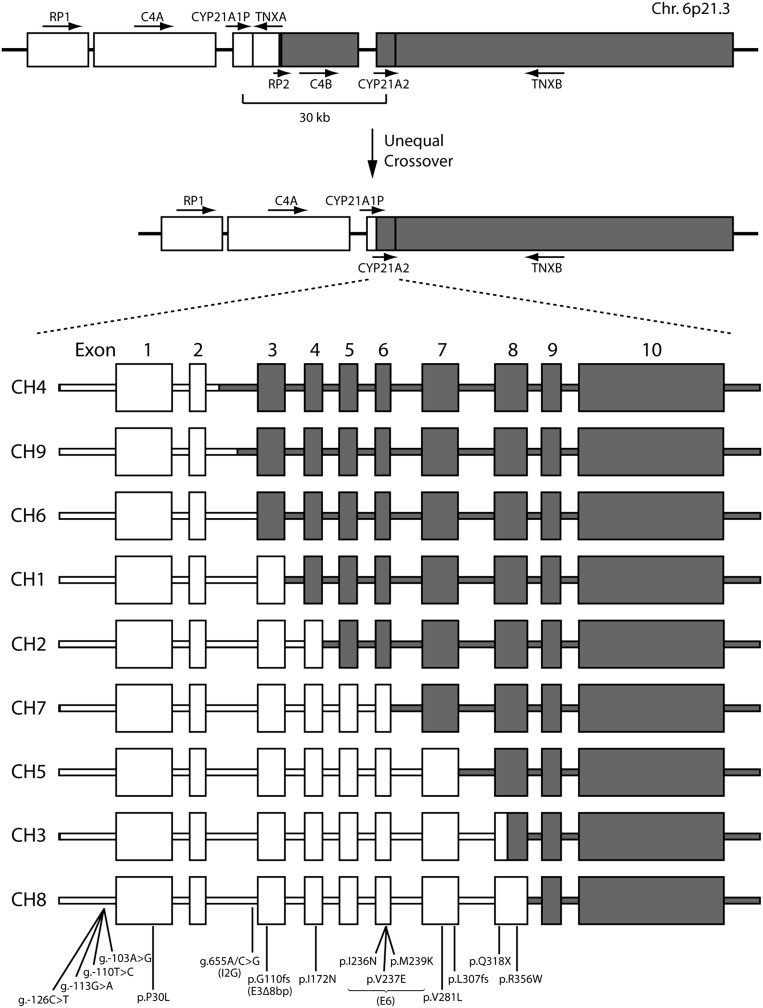
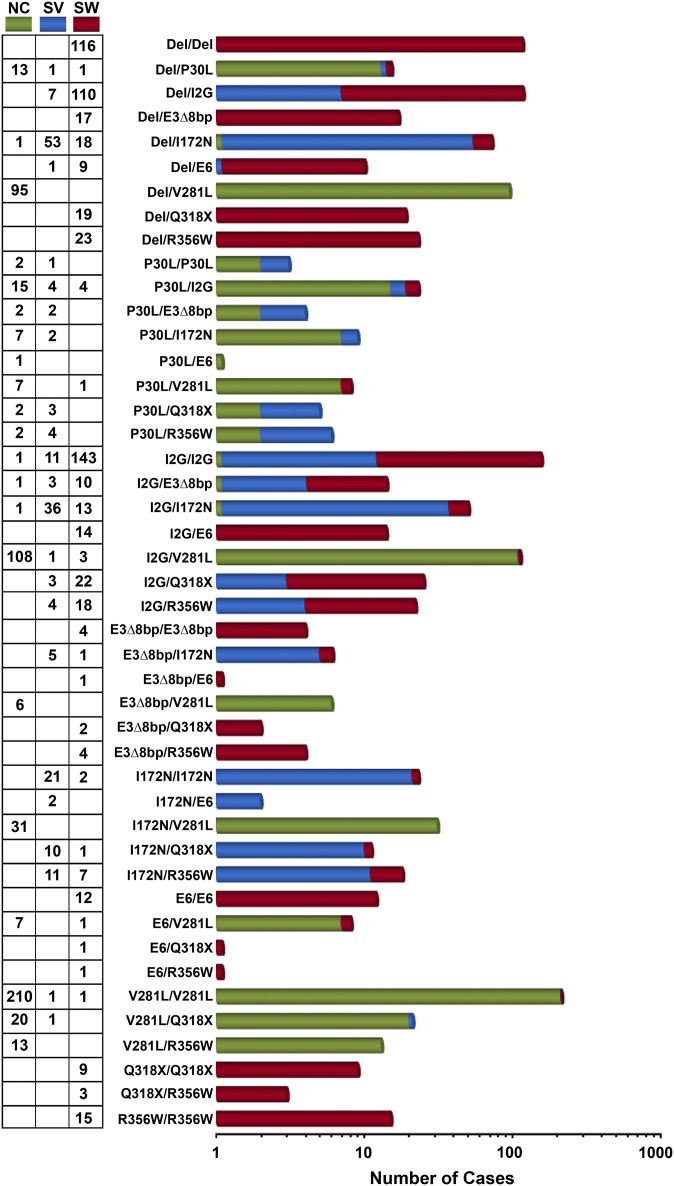
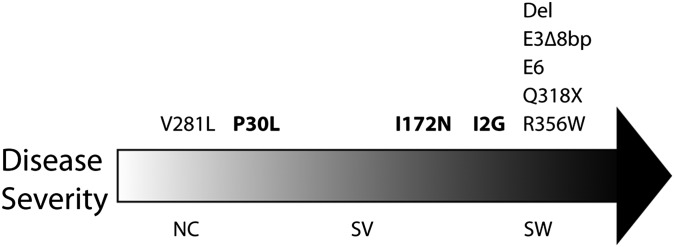
Over the last two decades, we have extensively studied the genetics of congenital adrenal hyperplasia caused by 21-hydroxylase deficiency (CAH) and have performed 8,290 DNA analyses of the CYP21A2 gene on members of 4,857 families at risk for CAH--the largest cohort of CAH patients reported to date. Of the families studied, 1,507 had at least one member affected with one of three known forms of CAH, namely salt wasting, simple virilizing, or nonclassical CAH. Here, we report the genotype and phenotype of each affected patient, as well as the ethnic group and country of origin for each patient. We showed that 21 of 45 genotypes yielded a phenotypic correlation in our patient cohort. In particular, contrary to what is generally reported in the literature, we found that certain mutations, for example, the P30L, I2G, and I172N mutations, yielded different CAH phenotypes. In salt wasting and nonclassical CAH, a phenotype can be attributed to a genotype; however, in simple virilizing CAH, we observe wide phenotypic variability, particularly with the exon 4 I172N mutation. Finally, there was a high frequency of homozygous I2G and V281L mutations in Middle Eastern and Ashkenazi Jewish populations, respectively. By identifying the predominant phenotype for a given genotype, these findings should assist physicians in prenatal diagnosis and genetic counseling of parents who are at risk for having a child with CAH.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Wajnrajch MP, New MI. Defects of adrenal steroidogenesis. In: Jameson JL, De Groot LJ, editors. Endocrinology. Philadelphia: Elsevier; 2010. 6th Ed, Vol 2, pp 1897–1920.
-
- Soardi FC, et al. Inhibition of CYP21A2 enzyme activity caused by novel missense mutations identified in Brazilian and Scandinavian patients. J Clin Endocrinol Metab. 2008;93(6):2416–2420. - PubMed
-
- Tardy V, et al. Phenotype-genotype correlations of 13 rare CYP21A2 mutations detected in 46 patients affected with 21-hydroxylase deficiency and in one carrier. J Clin Endocrinol Metab. 2010;95(3):1288–1300. - PubMed
-
- Chiou SH, Hu MC, Chung BC. A missense mutation at Ile172—Asn or Arg356—Trp causes steroid 21-hydroxylase deficiency. J Biol Chem. 1990;265(6):3549–3552. - PubMed
-
- Tusie-Luna MT, Traktman P, White PC. Determination of functional effects of mutations in the steroid 21-hydroxylase gene (CYP21) using recombinant vaccinia virus. J Biol Chem. 1990;265(34):20916–20922. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
