

Genomes of replicatively senescent cells undergo global epigenetic changes leading to gene silencing and activation of transposable elements
- PMID: 23360310
- PMCID: PMC3618682
- DOI: 10.1111/acel.12047
Genomes of replicatively senescent cells undergo global epigenetic changes leading to gene silencing and activation of transposable elements
Abstract
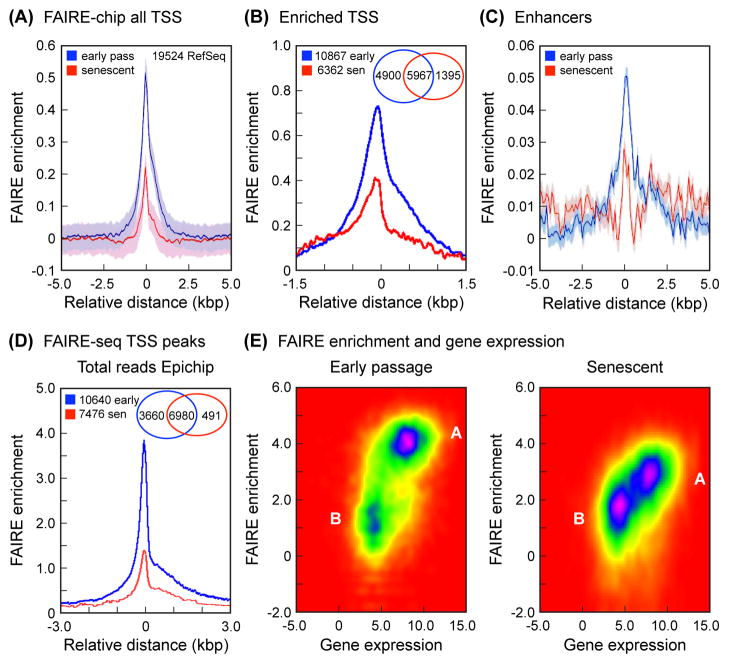
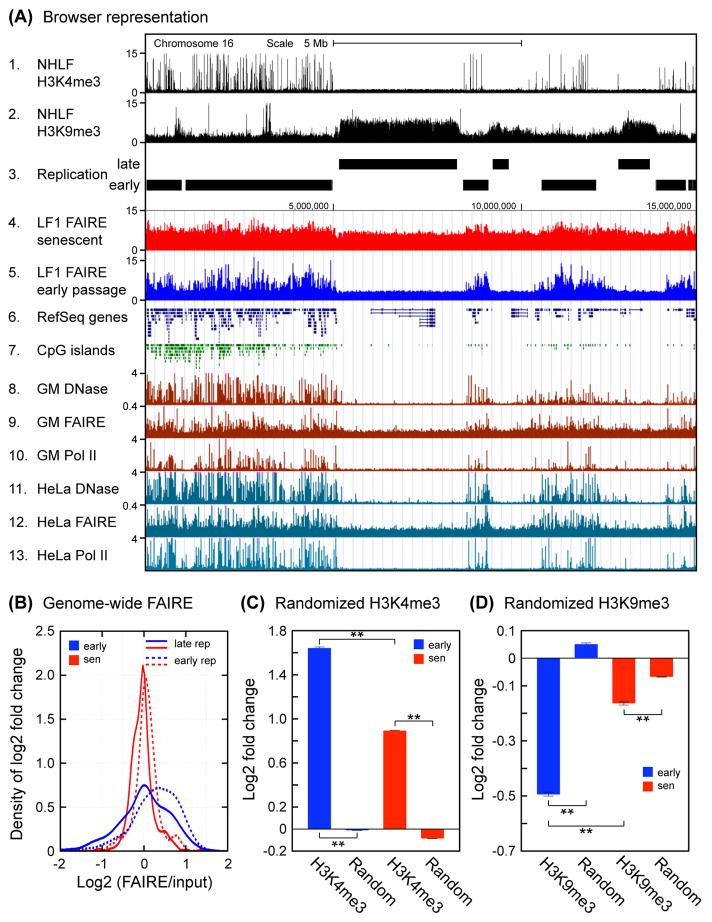
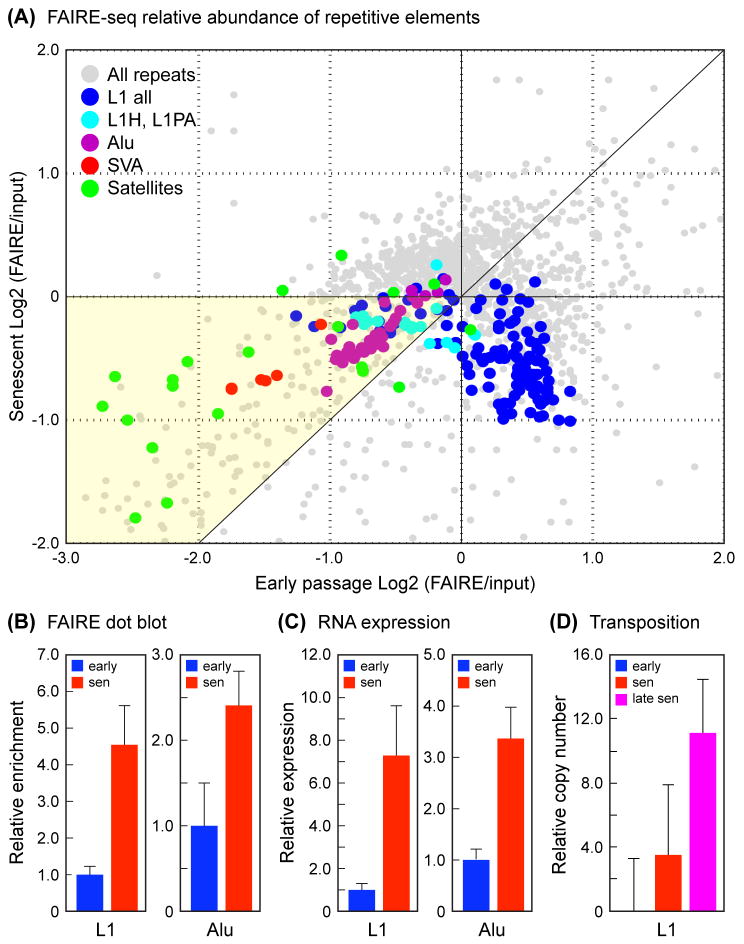
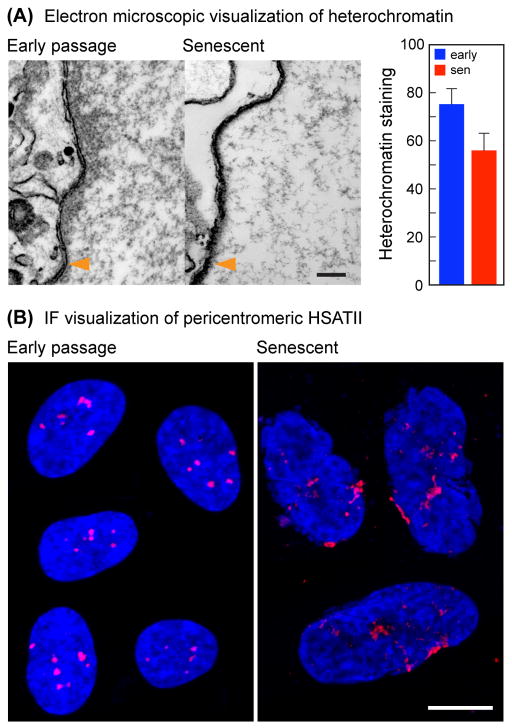
Replicative cellular senescence is an important tumor suppression mechanism and also contributes to aging. Progression of both cancer and aging include significant epigenetic components, but the chromatin changes that take place during cellular senescence are not known. We used formaldehyde assisted isolation of regulatory elements (FAIRE) to map genome-wide chromatin conformations. In contrast to growing cells, whose genomes are rich with features of both open and closed chromatin, FAIRE profiles of senescent cells are significantly smoothened. This is due to FAIRE signal loss in promoters and enhancers of active genes, and FAIRE signal gain in heterochromatic gene-poor regions. Chromatin of major retrotransposon classes, Alu, SVA and L1, becomes relatively more open in senescent cells, affecting most strongly the evolutionarily recent elements, and leads to an increase in their transcription and ultimately transposition. Constitutive heterochromatin in centromeric and peri-centromeric regions also becomes relatively more open, and the transcription of satellite sequences increases. The peripheral heterochromatic compartment (PHC) becomes less prominent, and centromere structure becomes notably enlarged. These epigenetic changes progress slowly after the onset of senescence, with some, such as mobilization of retrotransposable elements becoming prominent only at late times. Many of these changes have also been noted in cancer cells.
© 2013 The Authors Aging Cell © 2013 Blackwell Publishing Ltd/Anatomical Society of Great Britain and Ireland.
Figures
References
-
- Batzer MA, Deininger PL. Alu repeats and human genomic diversity. Nat Rev Genet. 2002;3:370–379. - PubMed
-
- Berman BP, Weisenberger DJ, Aman JF, Hinoue T, Ramjan Z, Liu Y, Noushmehr H, Lange CP, van Dijk CM, Tollenaar RA, Van Den Berg D, Laird PW. Regions of focal DNA hypermethylation and long-range hypomethylation in colorectal cancer coincide with nuclear lamina-associated domains. Nat Genet. 2012;44:40–46. - PMC - PubMed
-
- Bowman AW, Azzalini A. Applied smoothing techniques for data analysis. New York: Oxford University Press; 1997.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
